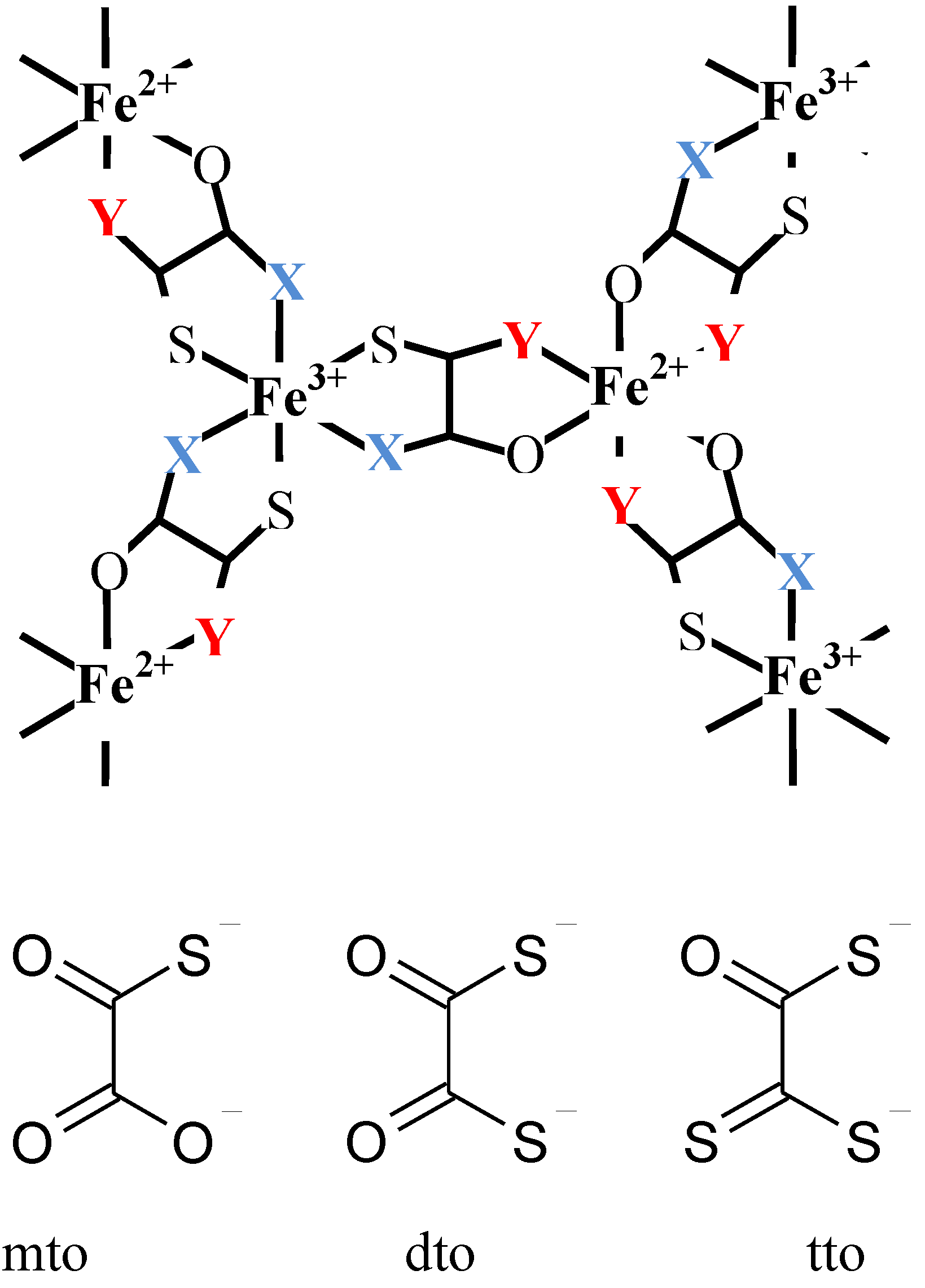
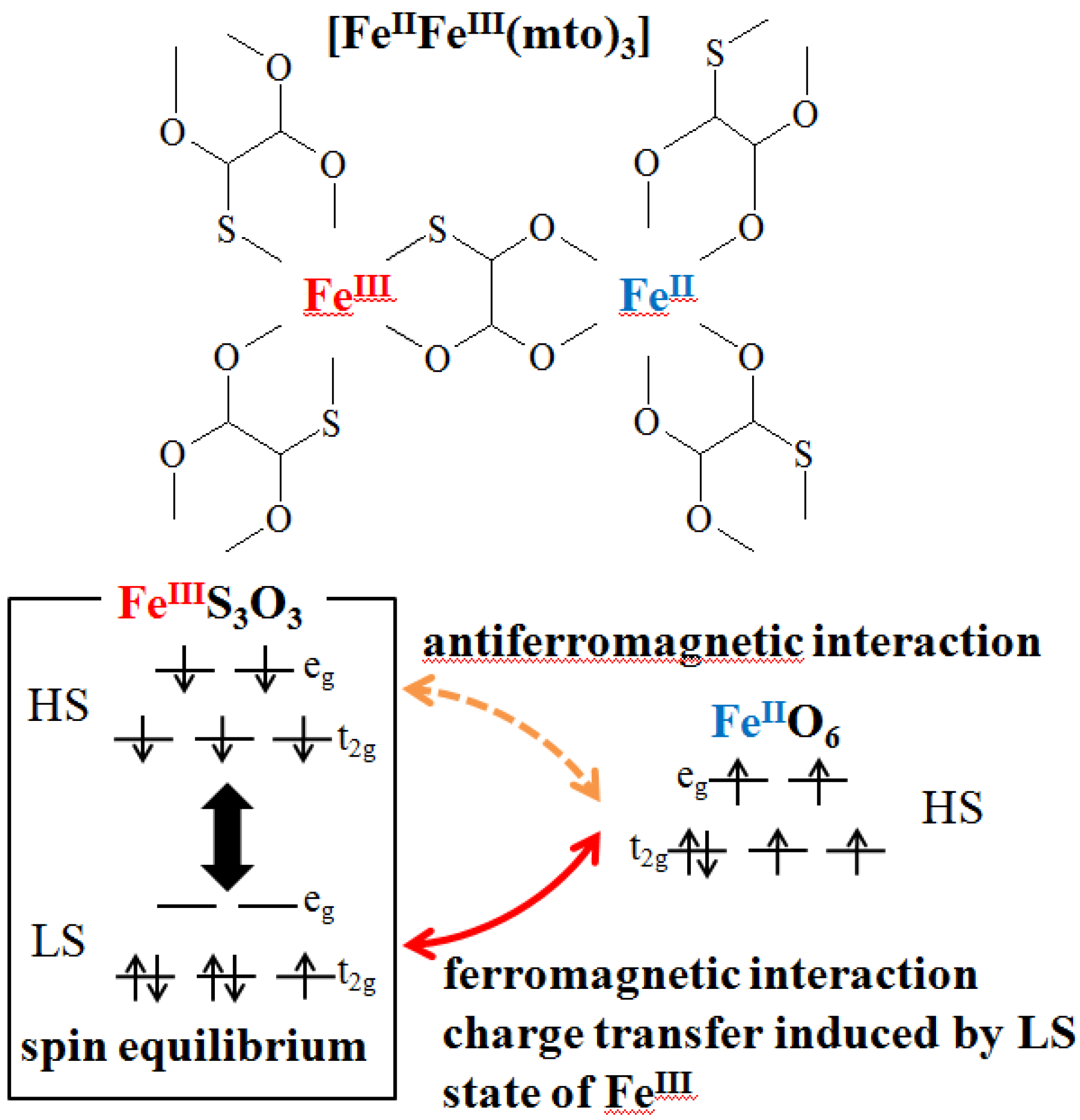
As mentioned in the chapter 1, the charge transfer phase transition (CTPT) is a new type of conjugated phenomenon coupled with spins and charges wherein a thermally induced metal-to-metal electron transfer occurs to minimize the free energy in the whole system [
33]. As a result of electron transfer, the spin degeneracy changes without spin transition, which is quite different from a classical spin crossover phenomenon. This spin-entropy effect has an important role in the driving force of the CTPT. In the cases of the iron mixed-valence complexes, (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] and (
n-C
4H
9)
4N[Fe
IIFe
III(dto)
3] show a novel CTPT between Fe
II and Fe
III in the [Fe
IIFe
III(dto)
3] layer. The existence of the CTPT depends on the size of cationic intercalant [
39,
40].
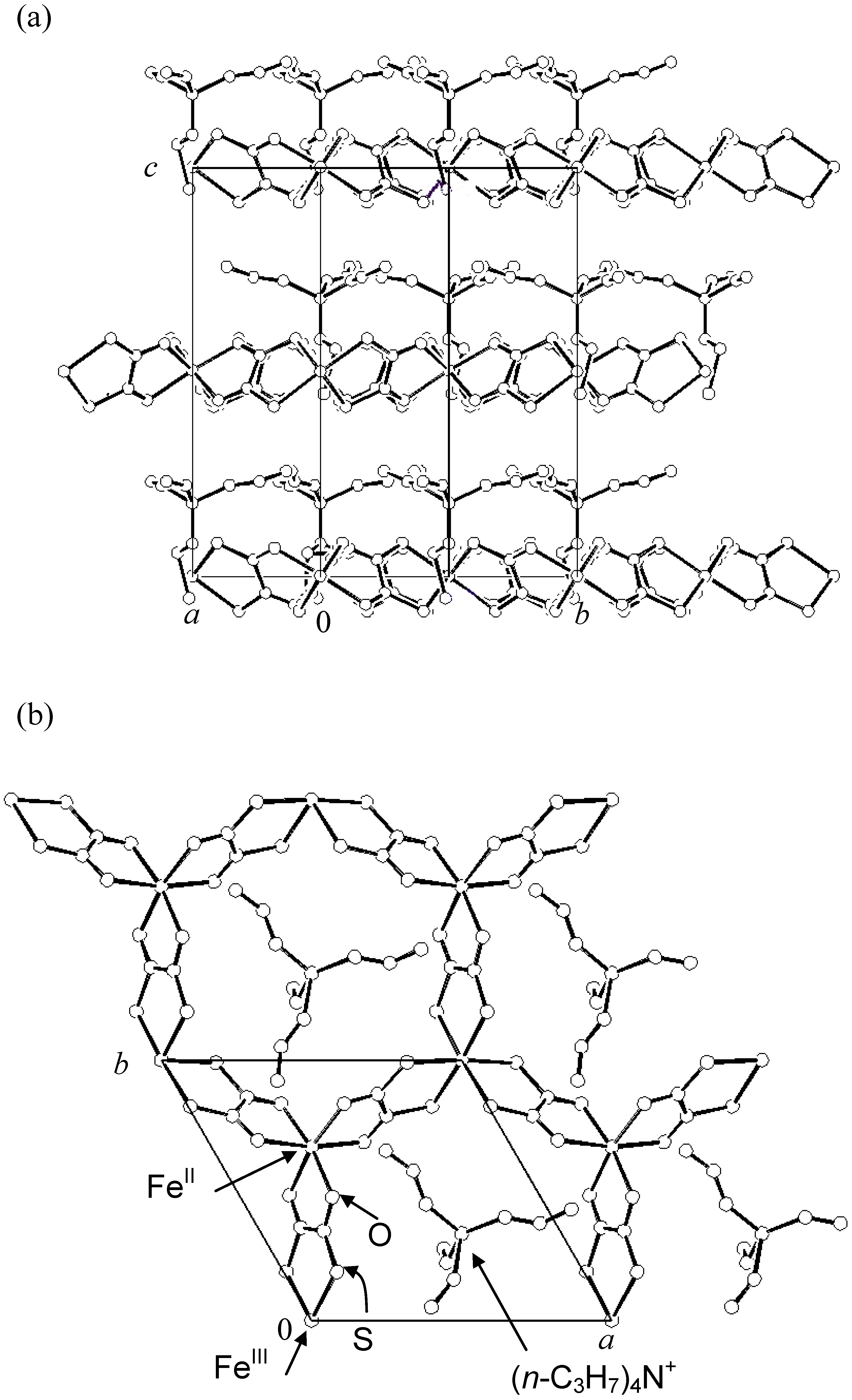
2.1. Crystal structure
Single crystals of (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] were grown by a diffusion method with the solvent of methanol–water mixture, where KBa[Fe
III(dto)
3]·3H
2O, FeCl
2·4H
2O were placed at one bottom of H-tube, while (
n-C
3H
7)
4NBr was kept at the other bottom. The single-crystal X-ray structural analysis reveals the existence of a two-dimensional (2D) honeycomb network structure of [Fe
IIFe
III(dto)
3] with intercalated counter cation as shown in
Figure 2 [
38]. The lattice parameters are as follows; space group
P6
3,
a = 10.0618(5) Å,
c = 16.0424(7) Å ,
V = 1406.54(12) Å
3,
Z = 2. The characteristic bond lengths of Fe-O and Fe-S for the as-prepared sample are in good agreement with those for Fe
II-O and Fe
III-S, respectively [
37,
54]. In this complex, the Fe
II and Fe
III atoms are alternately bridged by dithiooxalato molecules, which forms the 2D honeycomb network structure of [Fe
IIFe
III(dto)
3]. The (
n-C
3H
7)
4N cation layer is intercalated between two adjacent [Fe
IIFe
III(dto)
3] layers. This structure is common in hetero-metal oxalato and dithiooxalato complexes [
37,
55].
As mentioned above, Fe
II and Fe
III sites in (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] are coordinated by six O and six S atoms at room temperature, respectively. Moreover, the
57Fe Mössbauer spectroscopy specifies their spin states.
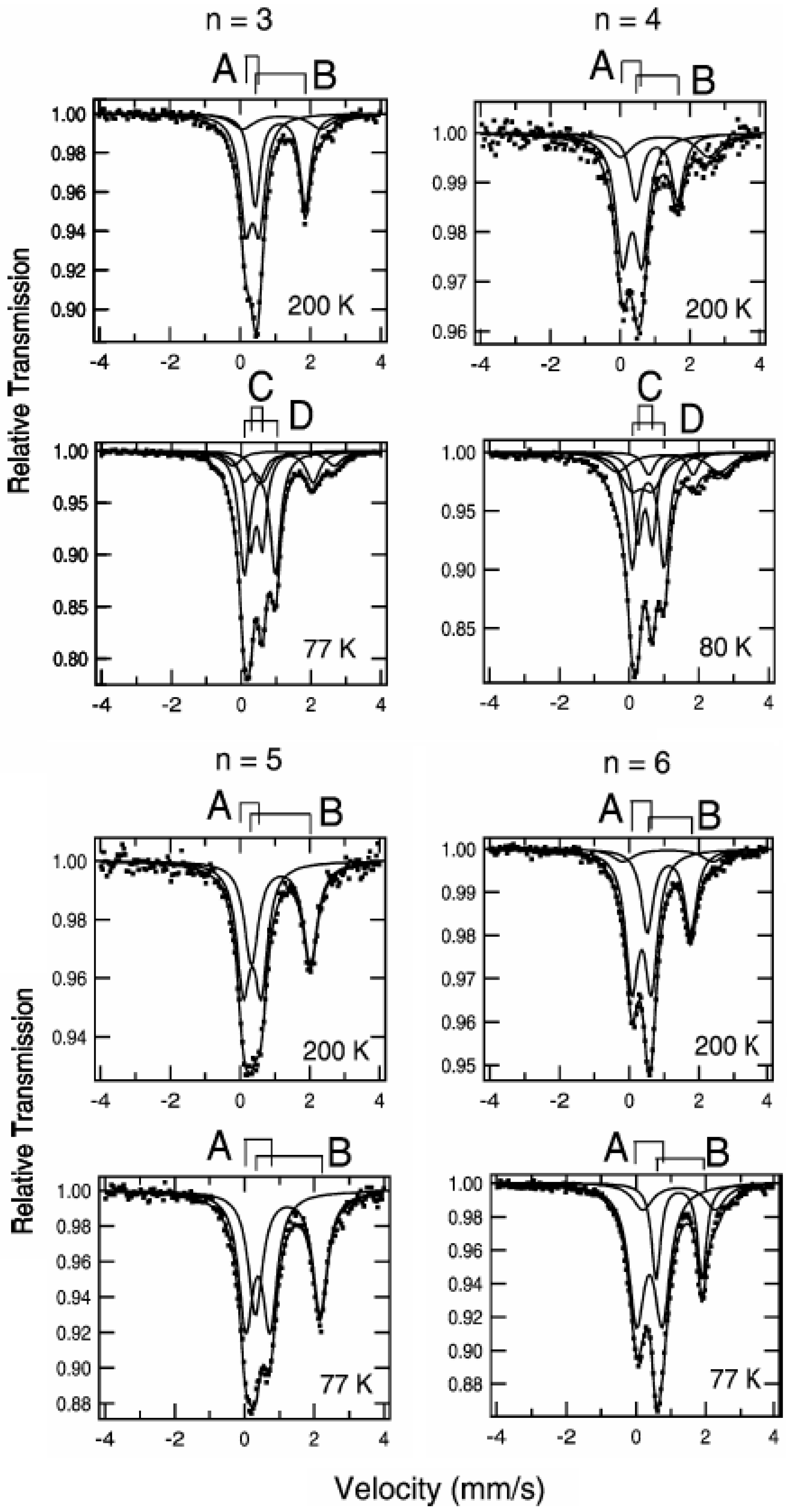
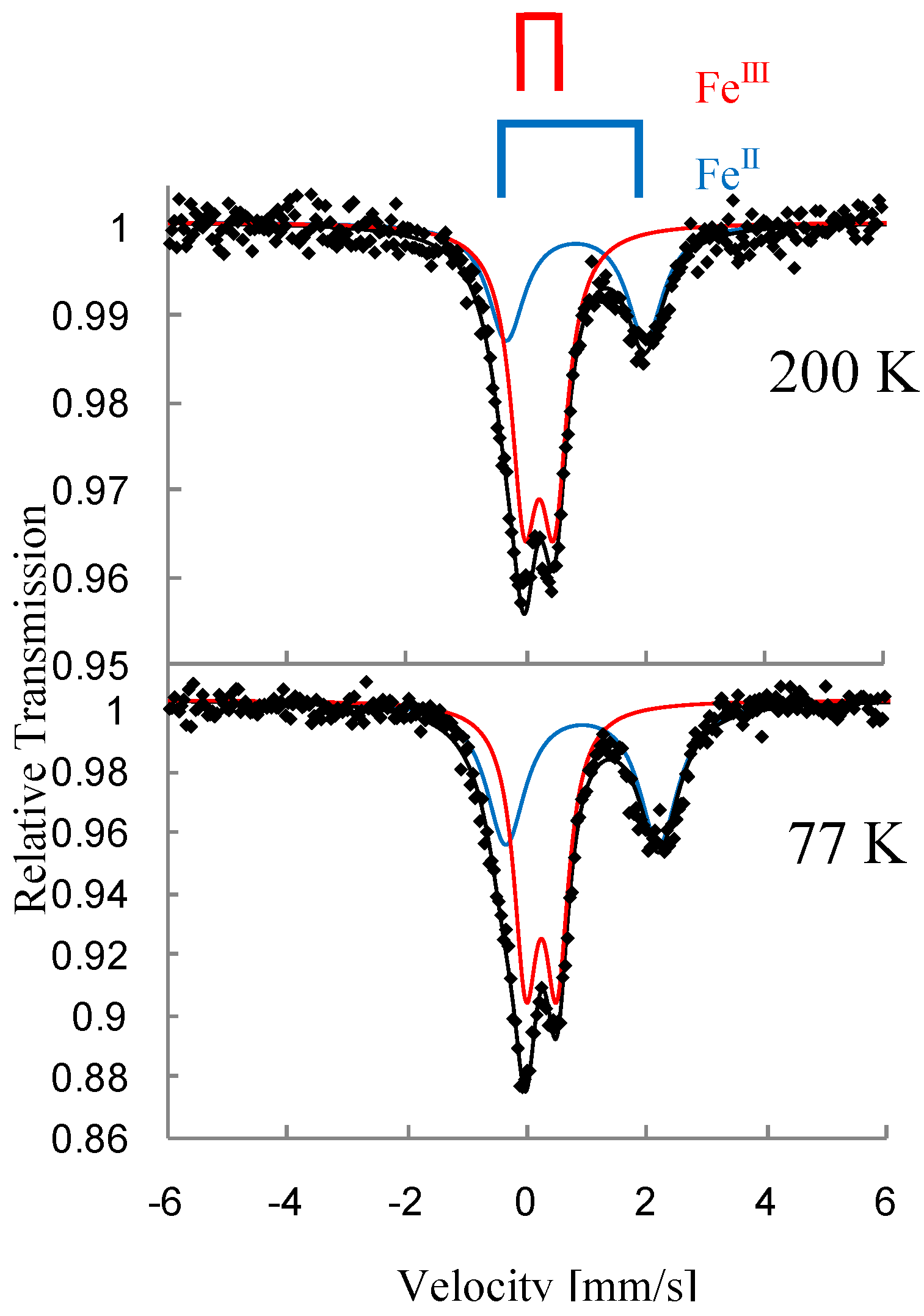
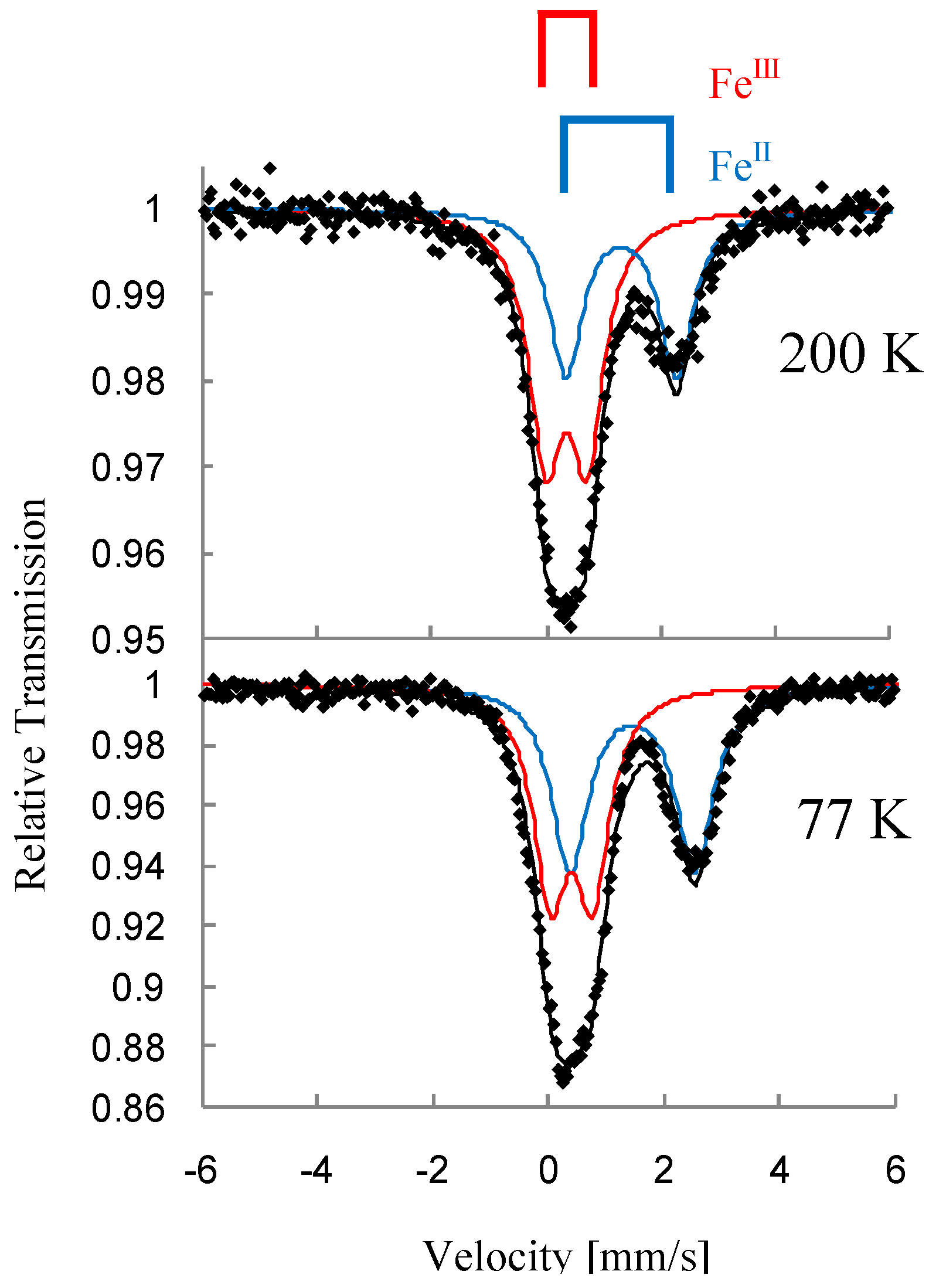
Figure 3 shows the
57Fe Mössbauer spectra for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3-6) [
39]. The assignment of the spectra A, B, C and D were confirmed by the
57Fe Mössbauer spectra of (
n-C
3H
7)
4N[
57Fe
IIFe
III(dto)
3] and (
n-C
3H
7)
4N[Fe
II 57Fe
III(dto)
3]. At 200 K, the line profiles of all the complexes are quite similar to each other. The isomer shift (
IS) and the quadrupole splitting (
QS) of the spectrum A at 200 K for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3-6) are quite similar to those (
IS = 0.33 mm/s,
QS = 0.35 mm/s at 196 K) of the
57Fe Mössbauer spectrum for the Fe
III(
S = 1/2) site in KBa[Fe
III(dto)
3]·3H
2O [
56], where the Fe
III site is coordinated by six S atoms. On the other hand, the
IS and
QS of the spectrum B at 200 K are quite similar to those (
IS = 1.235 mm/s,
QS = 1.42 mm/s at 190 K) of the
57Fe Mössbauer spectrum for the Fe
II(
S = 2) site in (
n-C
4H
9)
4N[Fe
IIFe
III(ox)
3](ox = oxalato) [
57], where the Fe
II site is coordinated by six O atoms. Therefore, it is concluded that the Fe
II(
S = 2) and Fe
III(
S = 1/2) sites in (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3-6) are coordinated by six O atoms and six S atoms, respectively. In the cases of n = 3, 4 and 6, besides the
57Fe Mössbauer spectra due to the Fe
IIIS
6 and Fe
IIO
6 octahedra, a weak spectrum with two branches at about 0.0 mm/s and 2.5 mm/s is observed, which is attributed to the high-spin state(
S = 2) of Fe
IIO
4S
2 octahedron caused by the linkage isomerization on precipitation process.
Figure 2.
Crystal structure of (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at room temperature.
(a) [210] projection,
(b) [001] projection [
38].
Figure 2.
Crystal structure of (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at room temperature.
(a) [210] projection,
(b) [001] projection [
38].
2.2. Charge transfer phase transition (CTPT) and ferromagnetism
In the cases of n = 3 and 4, at 77 K (80 K for n = 4), the spectra A and B decrease by about 80%. Instead of these spectra, the spectra C and D appear. The
IS and
QS of the spectrum C for n = 3 and 4 are similar to those (
IS = 0.325 mm/s,
QS = 0.39 mm/s) of the Fe
II(
S = 0) site in [Fe
II(bipy)
3](ClO
4)
2 (bipy = 2,2’-bipyridine) [
58]. On the other hand, the
IS and QS of the spectrum D for n = 3 and 4 are similar to those (
IS = 0.486,
QS = 0.64 at 90 K) of the
57Fe Mössbauer spectrum for the Fe
III(
S = 5/2) site in (
n-C
4H
9)
4N [Fe
IIFe
III(ox)
3] [
57].
As shown later in this section, n = 3 undergoes the ferromagnetic transition at 7 K [
32].
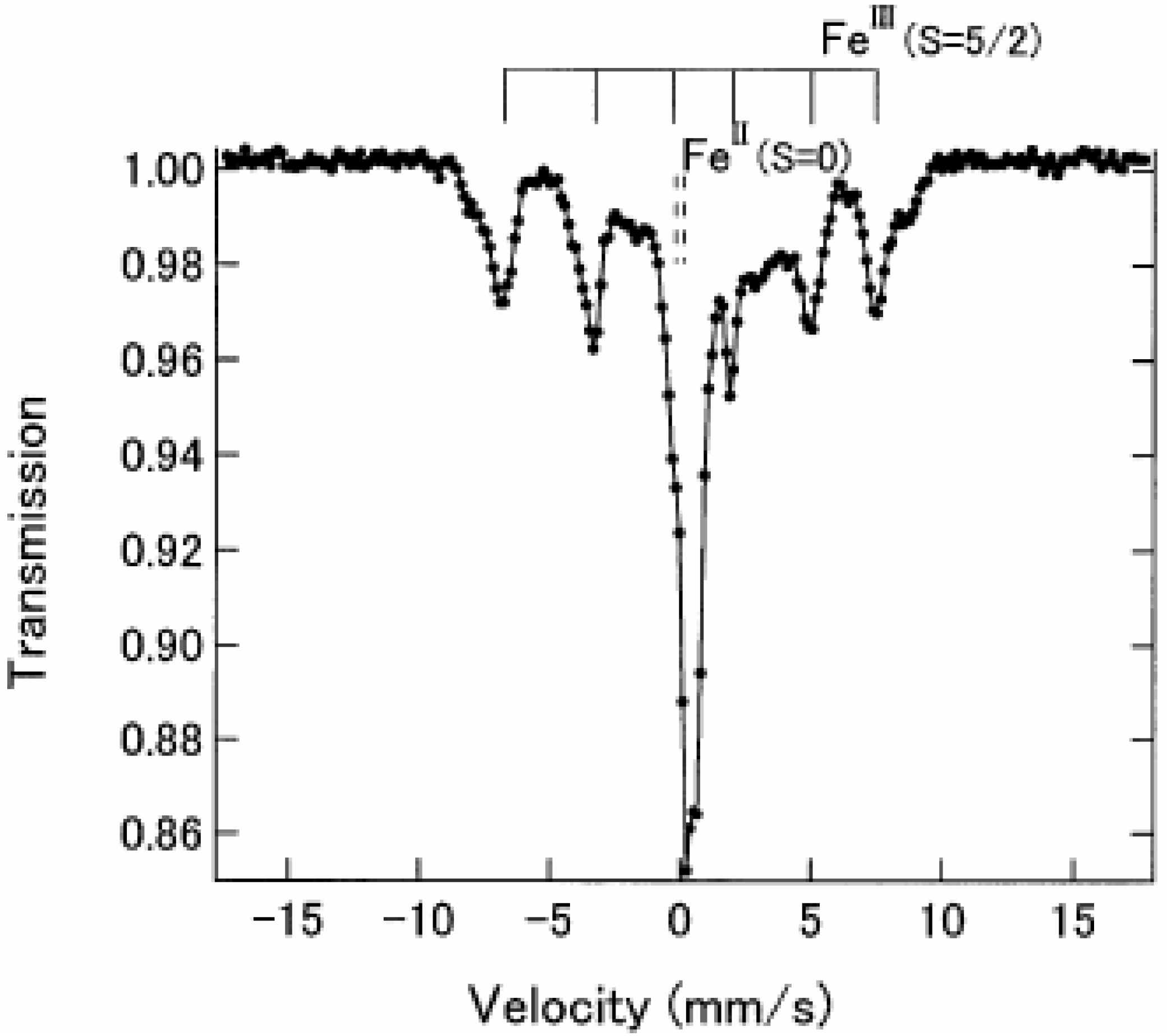
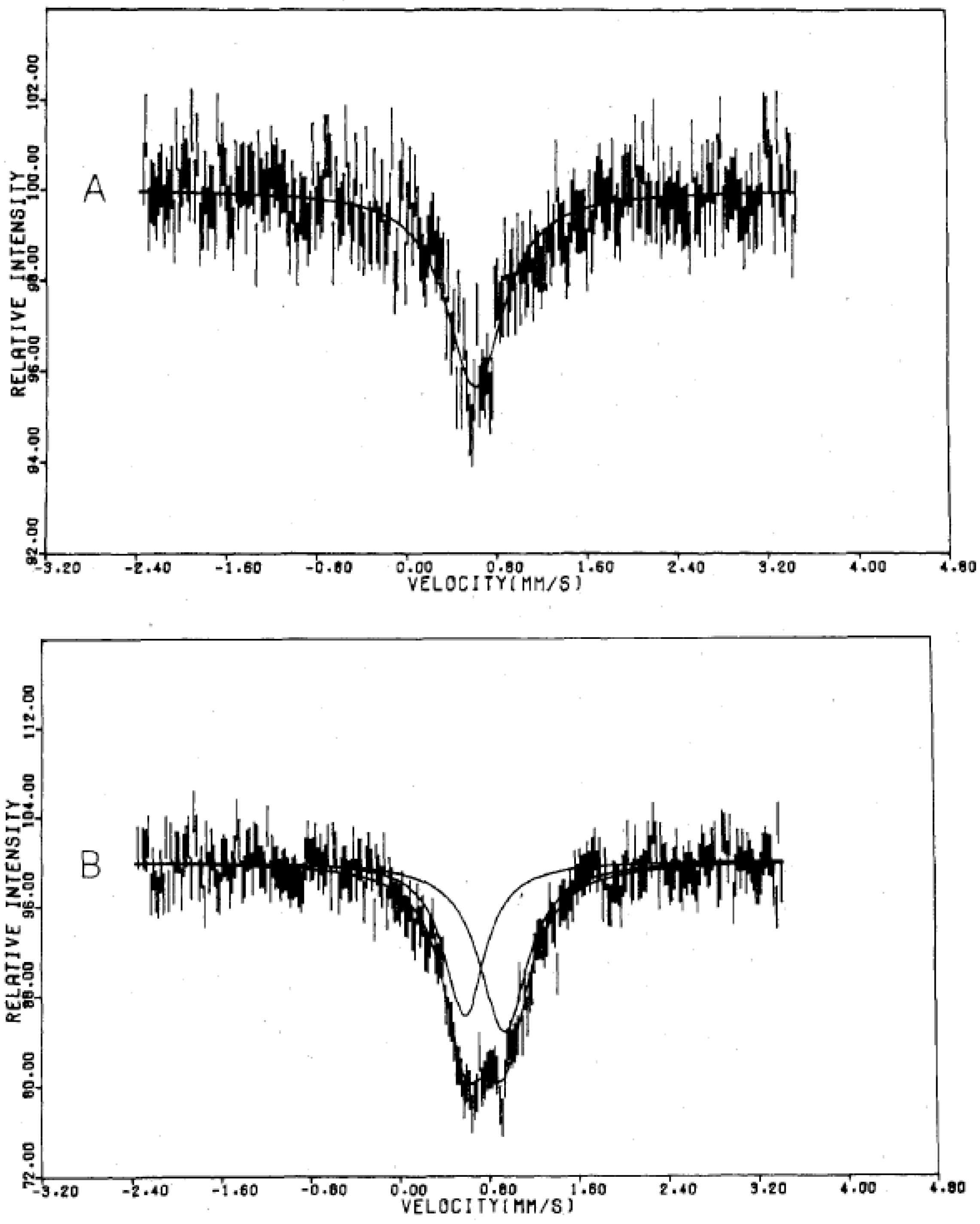
Figure 4 shows the
57Fe Mössbauer spectra in the ferromagnetically ordered phase of n = 3 [
32]. They are well resolved and show a superposition of a central peak and a magnetically split spectrum with six branches. The line profile of
57Fe Mössbauer spectrum for n = 3 in the ferromagnetically ordered phase is quite similar to that of the
57Fe Mössbauer spectrum for Prussian blue, (Fe
III[Fe
II(CN)
6]
3) below
TC ( = 5.5 K) [
59]. In the case of Prussian blue, the spin state of the Fe
II site coordinated by six C atoms is the low spin state (
S = 0), and that of the Fe
III site coordinated by N atoms is the high spin state (
S = 5/2). The estimated internal magnetic fields for Fe
II (
S = 0) and Fe
III (
S = 5/2) in Fe
III[Fe
II(CN)
6]
3 at 1.6 K are 0 and 540 kOe, respectively [
59]. Comparing the internal magnetic fields for the Fe
II and Fe
III sites in n = 3, the
57Fe Mössbauer spectrum with six split branches induced by the internal magnetic field of 446 kOe corresponds to that for the Fe
III site with high spin state (
S = 5/2), and the central peak without internal magnetic field corresponds to that for the Fe
II site with low spin state (
S = 0). Taking into account the obtained
IS and
QS of the
57Fe Mössbauer spectra for the Fe
II and Fe
III sites at 4.2 K, the
57Fe Mössbauer spectra of n = 3 at 77 K can be reproduced as shown in
Figure 3.
From the
57Fe Mössbauer spectra for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3 and 4), it is obvious that the charge transfer phase transition (CTPT) takes place between 200 K and 77 K for n = 3 and 4. The coexistence of the higher and lower temperature phases at 77 K is typical of first order phase transition, which reflects on the thermal hysteresis in magnetic susceptibility [
40]. From the analysis of heat capacity, the critical temperature of the CTPT was determined at 122.4 K for n = 3 [
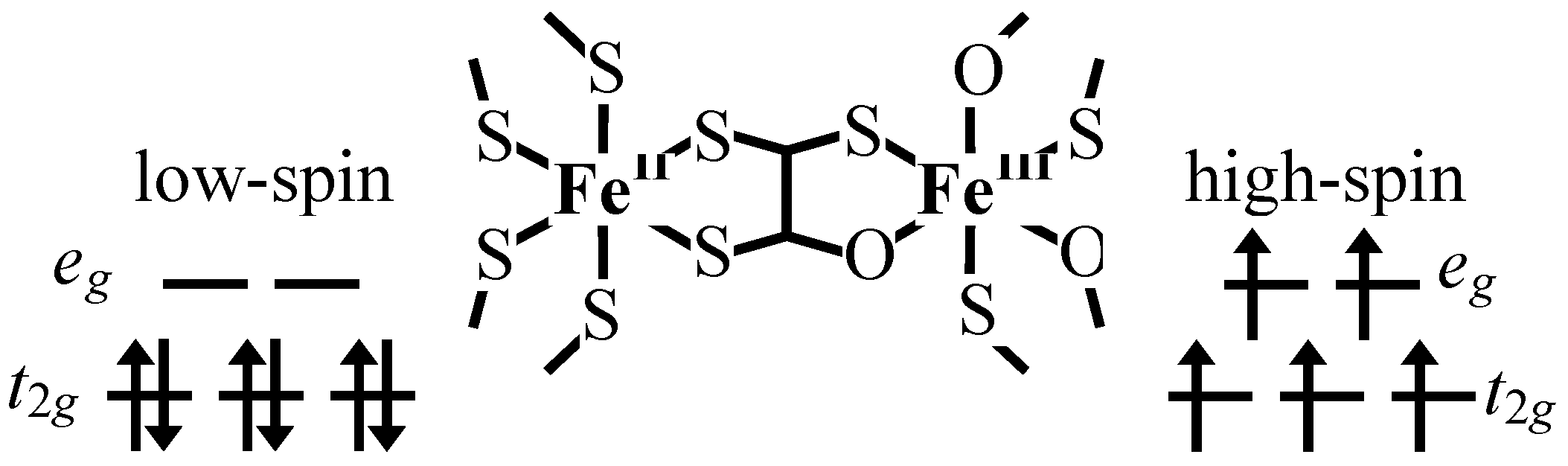
33]. In the high temperature phase, the spin configuration becomes Fe
II(S = 2)-Fe
III(S = 1/2) for as-prepared sample. Below 120 K, on the other hand, the spin state of the Fe
II coordinated by six S atoms should be
S = 0 and that of the Fe
III coordinated by six O atoms should be
S = 5/2, which is able to explain the values of
IS,
QS and
Hint of the
57Fe Mössbauer spectra in the low temperature phase.
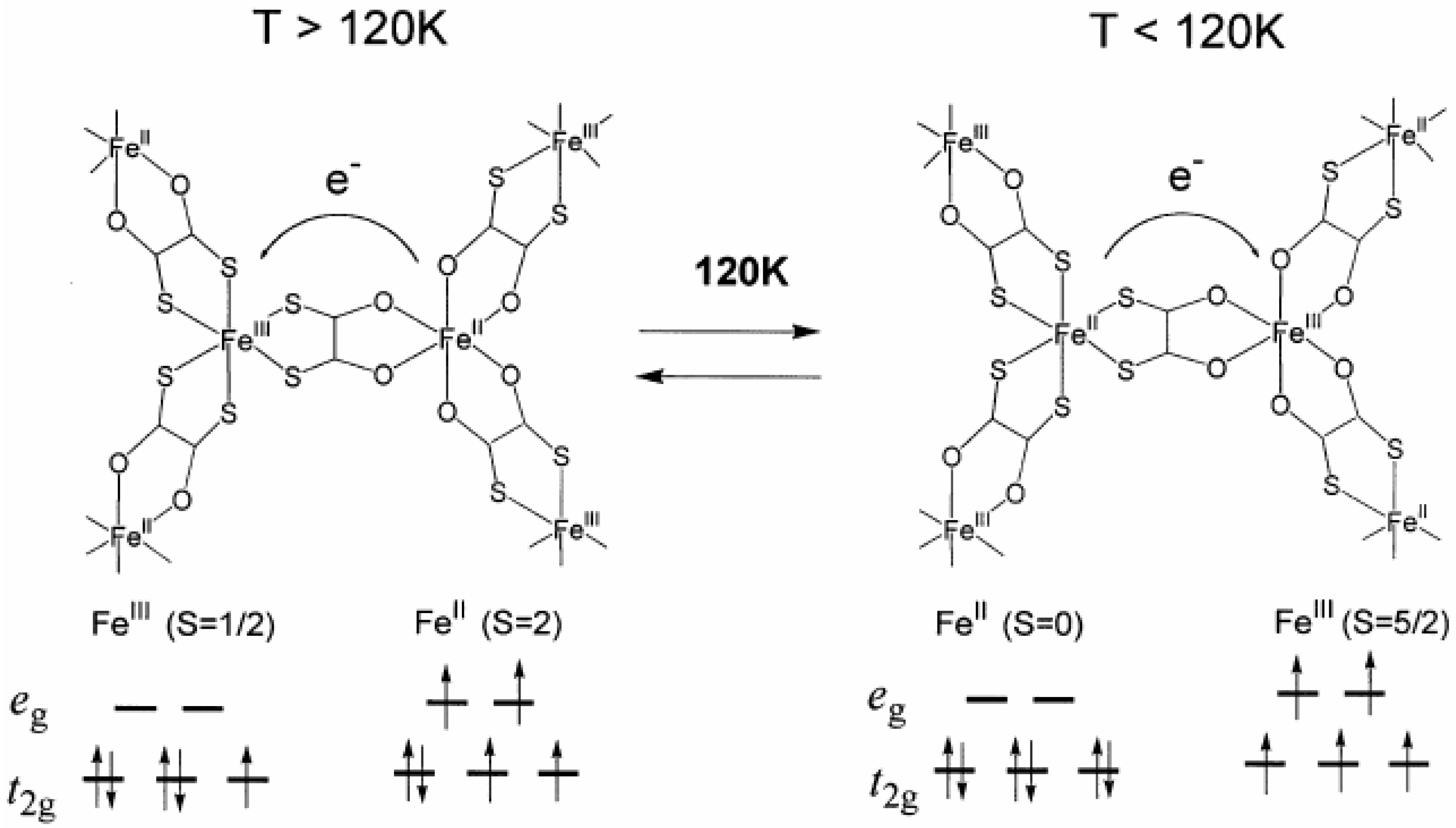
Therefore, it is concluded that (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] undergoes a thermally induced CTPT at about 120 K where the electron transfer occurs reversibly between the
t2g orbitals of the Fe
II and Fe
III sites, which is schematically shown in
Figure 5. The driving force responsible for the CTPT is the difference in spin entropy between the high temperature phase (HTP) and the low temperature one (LTP). It should be noted that the spin entropy in the high temperature phase is R ln(2 × 5) = 19.15 J·K
-1·mol
-1and that in the low temperature phase is R ln(1 × 6) = 14.90 J·K
-1·mol
-1, where R is the gas constant. Therefore, the spin-entropy gain expected from the electron transfer from the
t2g orbital of the Fe
II site to that of the Fe
III site is estimated at 4.25 J·K
-1·mol
-1. Since the observed entropy gain at the CTPT in (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] is 9.20 J·K
-1·mol
-1 [
33], the entropy change originating from the lattice vibration is quite smaller than that for classical spin-crossover transition. For example, about 35 J·K
-1·mol
-1 was estimated for the vibrational contribution to the entropy change in the spin crossover transition in [Fe(phen)
2(NCS)
2] [
60].
Figure 3.
57Fe Mössbauer spectra for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3 – 6). A: Fe
III(
S = 1/2), B: Fe
II(
S = 2), C: Fe
II(
S = 0), D: Fe
III(
S = 5/2) [
39].
Figure 3.
57Fe Mössbauer spectra for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3 – 6). A: Fe
III(
S = 1/2), B: Fe
II(
S = 2), C: Fe
II(
S = 0), D: Fe
III(
S = 5/2) [
39].
Figure 4.
57Fe Mössbauer spectrum of (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at 4.2 K. Solid and broken lines show the spectral peak positions of the
57Fe Mössbauer spectrum corresponding to the Fe
III(
S = 5/2) and Fe
II (
S = 0) sites, respectively [
32].
Figure 4.
57Fe Mössbauer spectrum of (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at 4.2 K. Solid and broken lines show the spectral peak positions of the
57Fe Mössbauer spectrum corresponding to the Fe
III(
S = 5/2) and Fe
II (
S = 0) sites, respectively [
32].
Figure 5.
Schematic feature of the charge transfer phase transition in (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] [
32].
Figure 5.
Schematic feature of the charge transfer phase transition in (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] [
32].
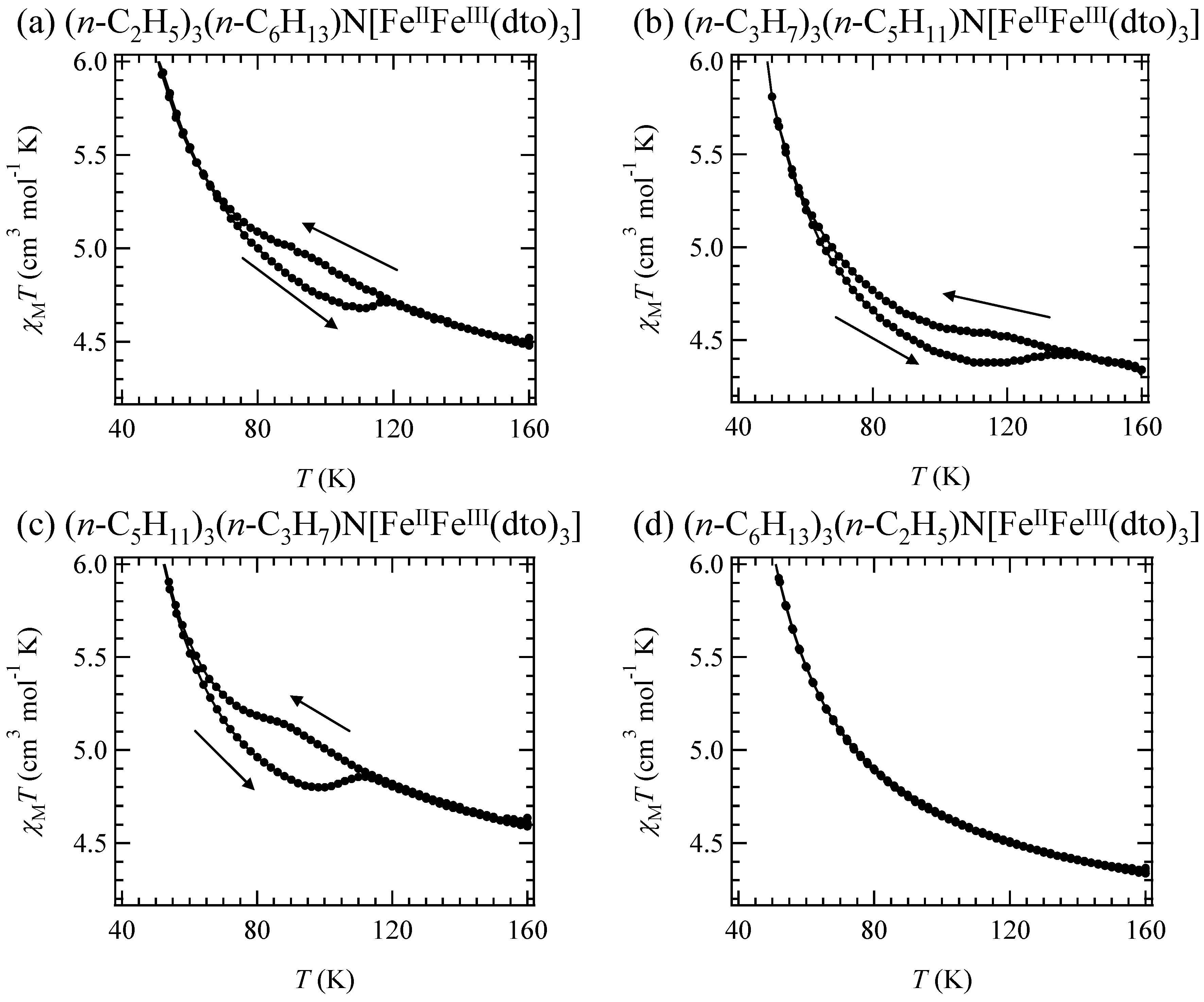
In the cases of n = 5 and 6, the line profile of the 57Fe Mössbauer spectra remains unchanged between 200 K and 77 K, which implies that the charge transfer phase transition does not take place for n = 5 and 6. In fact, the higher temperature phase exists between 300 K and 4 K for n = 5 and 6. The charge transfer phase transition is sensitive to the size of 2D honeycomb network structure. The increase of cation size expands the honeycomb ring, which presumably stabilizes the higher temperature phase.
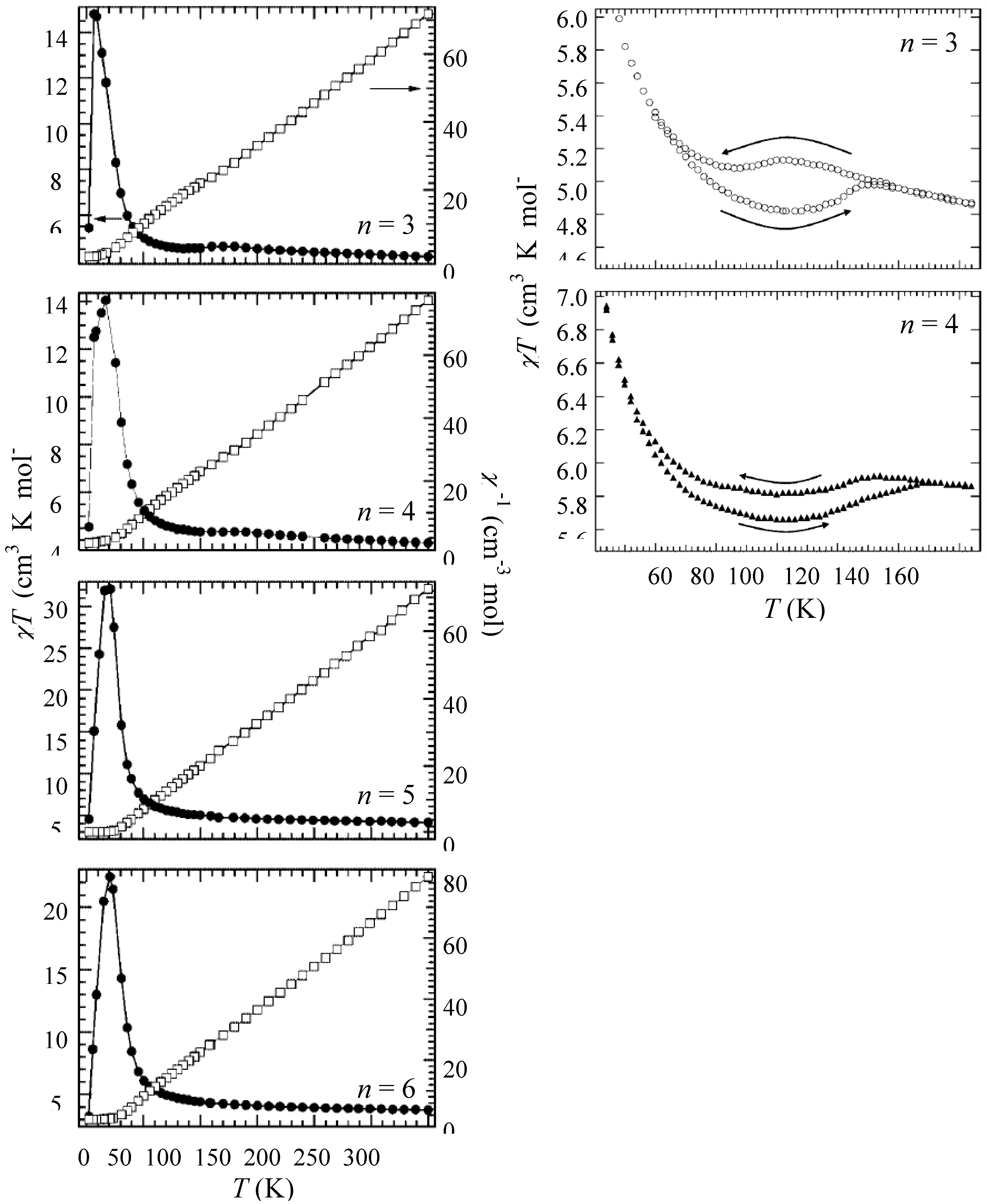
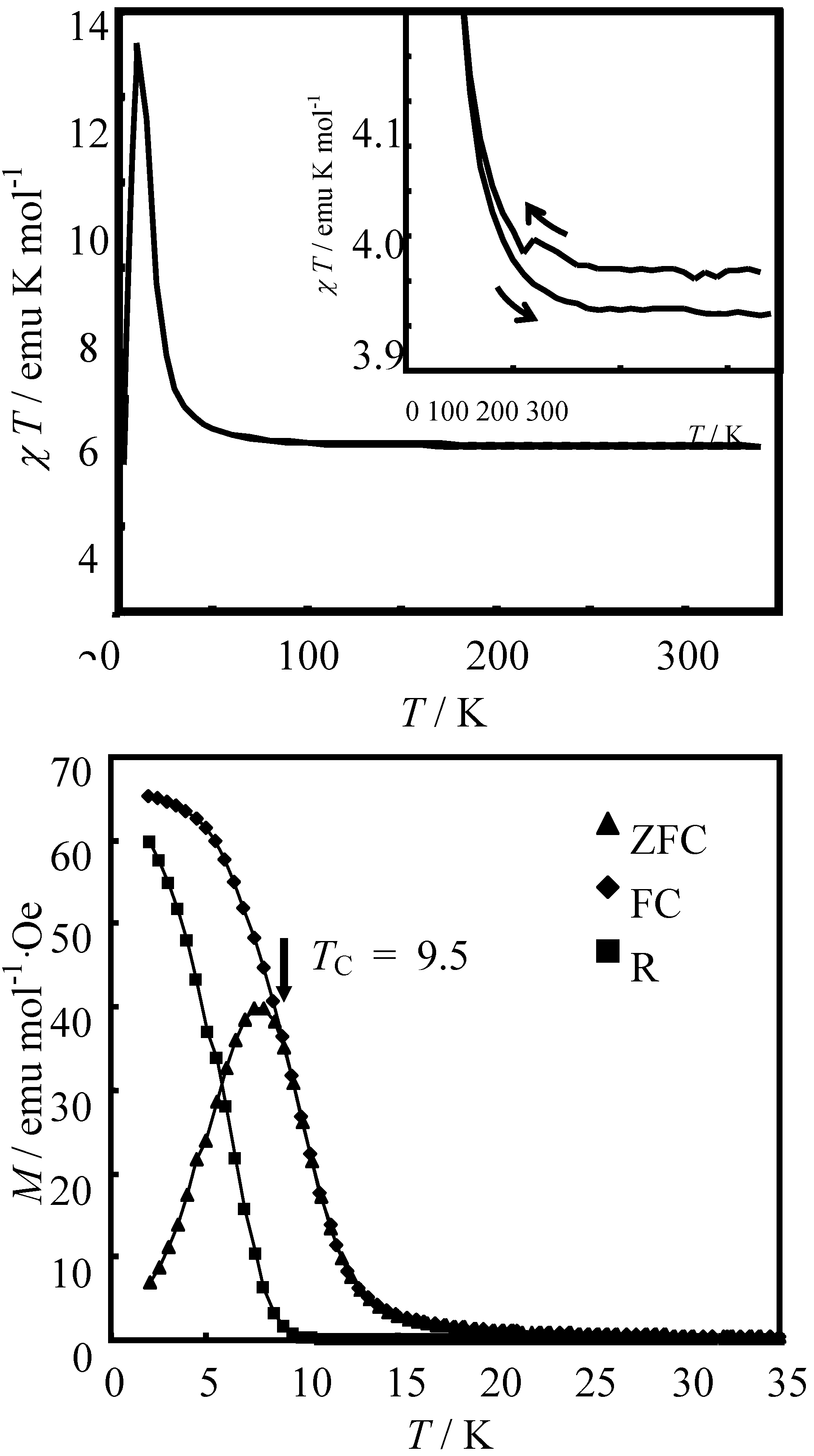
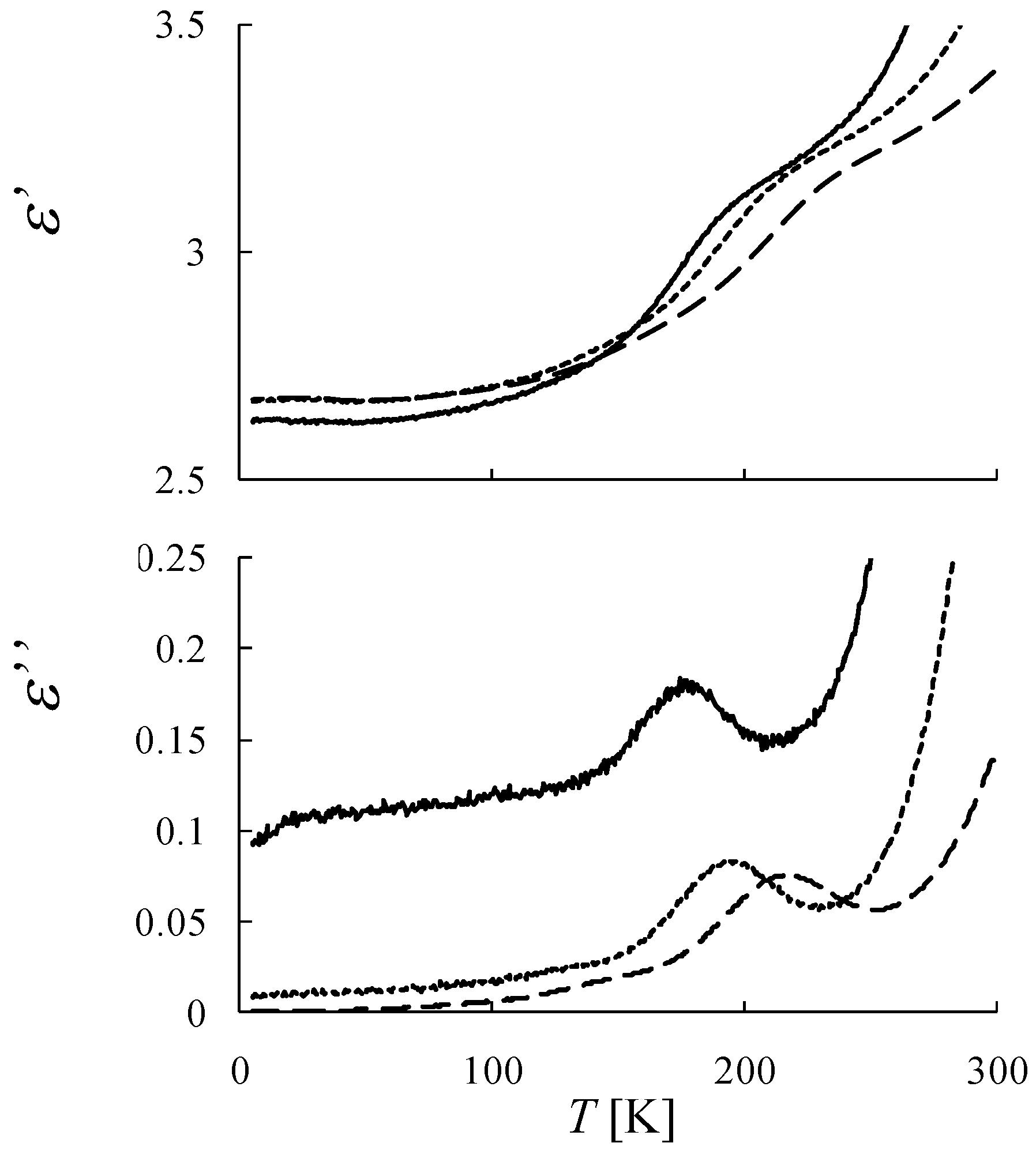
Figure 6 shows the χ
T and the inverse magnetic susceptibility (χ
–1) as a function of temperature for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3-6) [
40]. The magnetic susceptibilities for all the complexes obey the Curie-Weiss law, χ
–1 = (
T-θ) /
T, in the range of 150-300 K. The Weiss constants of n = 3 – 6 are estimated at +12 K, +18 K, +23 K and +21 K, respectively. All the positive Weiss constants imply the ferromagnetic interaction between Fe
II and Fe
III in (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3]. In the cases of n = 3 and 4, reflecting the CTPT, a small bump appears in the χ
T curve and the slope of χ
–1 changes around 120 K and 140 K for n = 3 and n = 4, respectively. In this temperature region, a thermal hysteresis loop appears between 60-130 K and 50-150 K for n = 3 and n = 4, respectively.
Figure 6.
Temperature dependence of the magnetic susceptibility multiplied by temperature (χ
T: ●) and the inverse susceptibility (χ
–1: ○) of (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3 – 6) [
40].
Figure 6.
Temperature dependence of the magnetic susceptibility multiplied by temperature (χ
T: ●) and the inverse susceptibility (χ
–1: ○) of (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3 – 6) [
40].
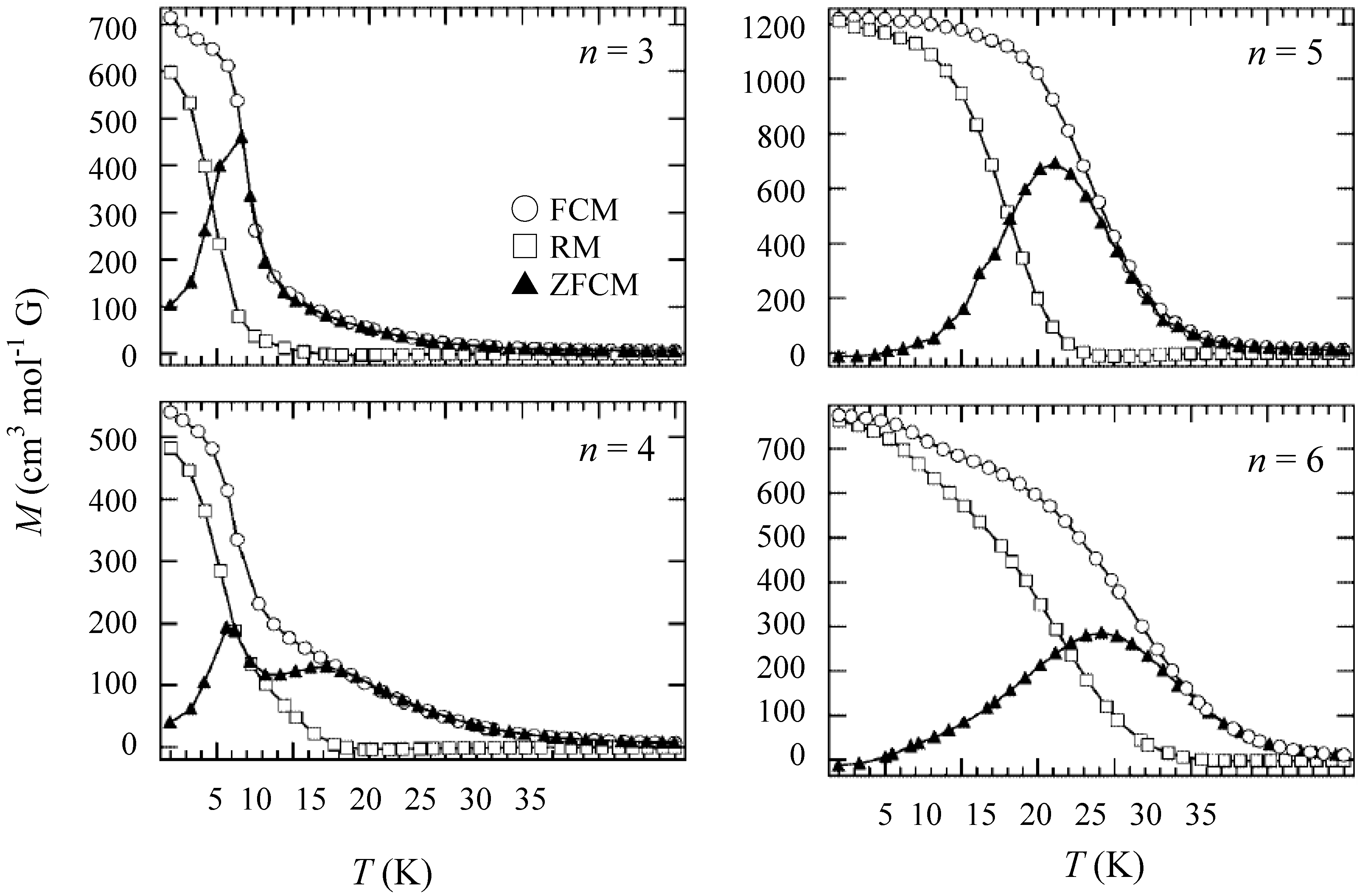
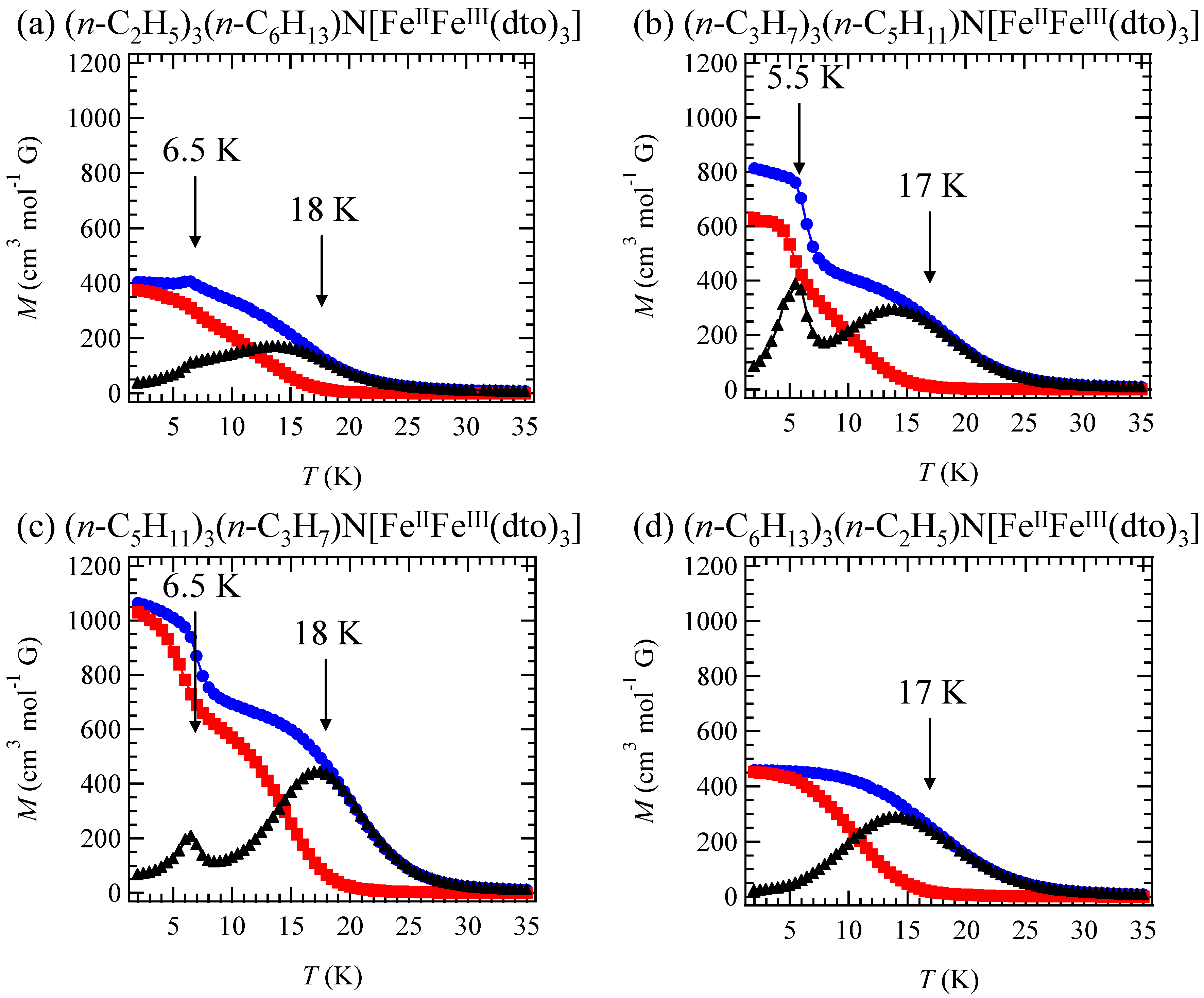
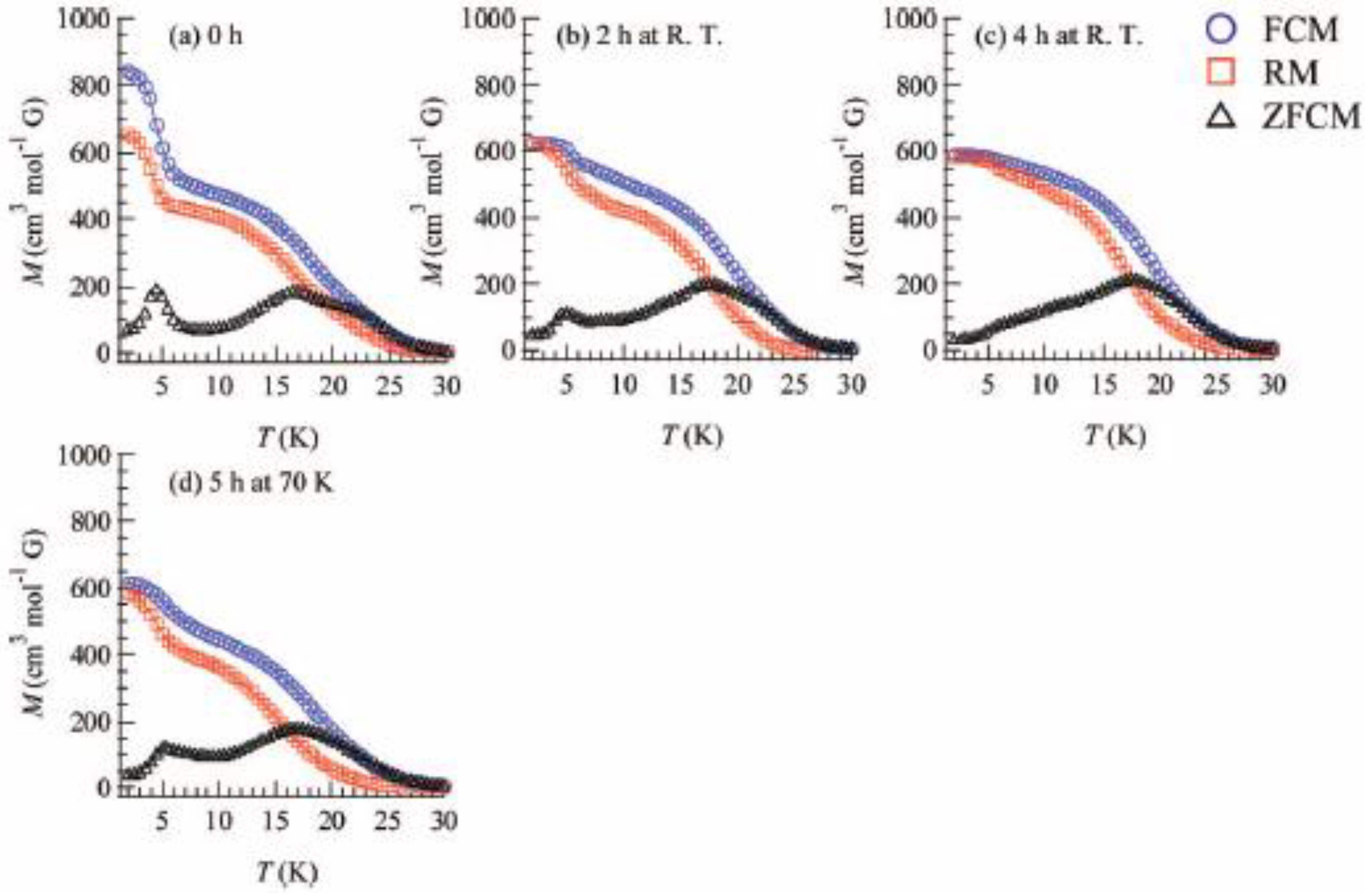
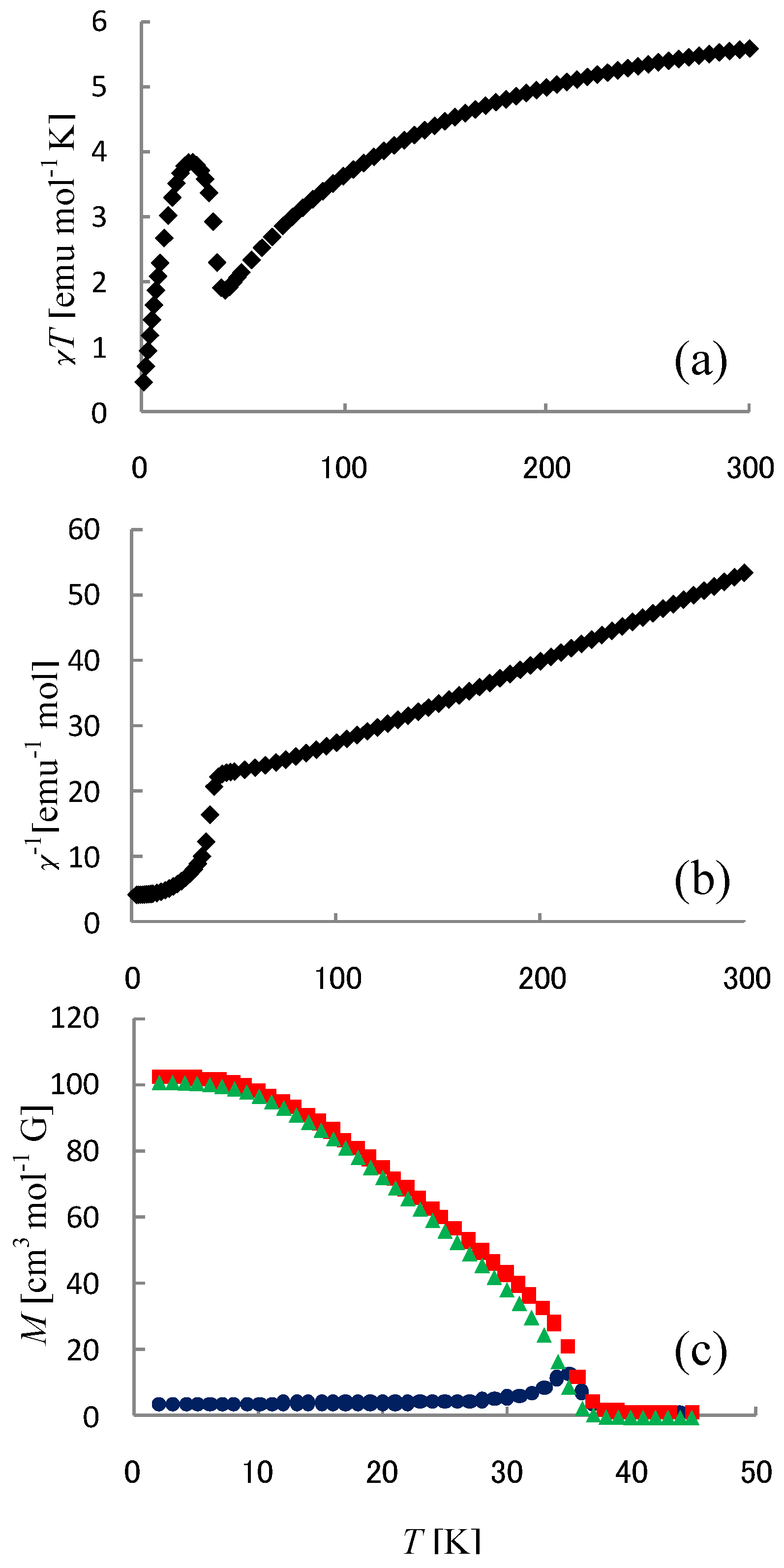
In order to confirm the ferromagnetic phase transition, we have investigated the field-cooled magnetization (FCM), the remnant magnetization (RM) and the zero-field cooled magnetization (ZFCM) for n = 3–6, which are shown in
Figure 7 [
40]. The FCM curve was obtained on cooling with an external magnetic field of 30 Oe. After the measurement of FCM, the magnetic field was switched off at 2 K and then the RM was measured from 2 K to 35 K. After cooling from 300 K to 2 K with complete zero external field, the external field of 30 Oe was switched on at 2 K, then the ZFCM was measured from 2 K to 35 K. The RM corresponds to the spontaneous magnetization. Below the ferromagnetic transition, the ZFCM is smaller than the FCM, which is due to the fact that the applied magnetic field of 30 Oe is too weak to move the magnetic domain walls below the Curie temperature. The ZFCM and FCM curves meet each other at the Curie temperature where the hysteresis disappears. In this way, the Curie temperature is evaluated. More precisely, the Curie temperature were estimated at 7, (7 and 13), 19.5 and 23 K for n =3-6, respectively, from the heat capacity measurements for n = 3 and 4, [
33] and from the analysis of Arrott plot for n = 5 and 6 [
40]. In the case of n = 4, the ZFCM has two peaks at 7 K and 13 K, and the RM vanishes at 13 K, which implies that the LTP and HTP coexist even at 2 K and these phases undergo the ferromagnetic phase transitions at
TC = 7 K and
TC = 13 K, respectively. The critical temperature of CTPT, the Weiss constant and the Curie temperature for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3-6) are summarized in
Figure 8.
Figure 7.
Temperature dependence of the field cooled magnetization (FCM), the remnant magnetization (RM), and the zero-field cooled magnetization (ZFCM) for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3–6). Applied magnetic field,
H = 30 Oe [
40].
Figure 7.
Temperature dependence of the field cooled magnetization (FCM), the remnant magnetization (RM), and the zero-field cooled magnetization (ZFCM) for (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3](n = 3–6). Applied magnetic field,
H = 30 Oe [
40].
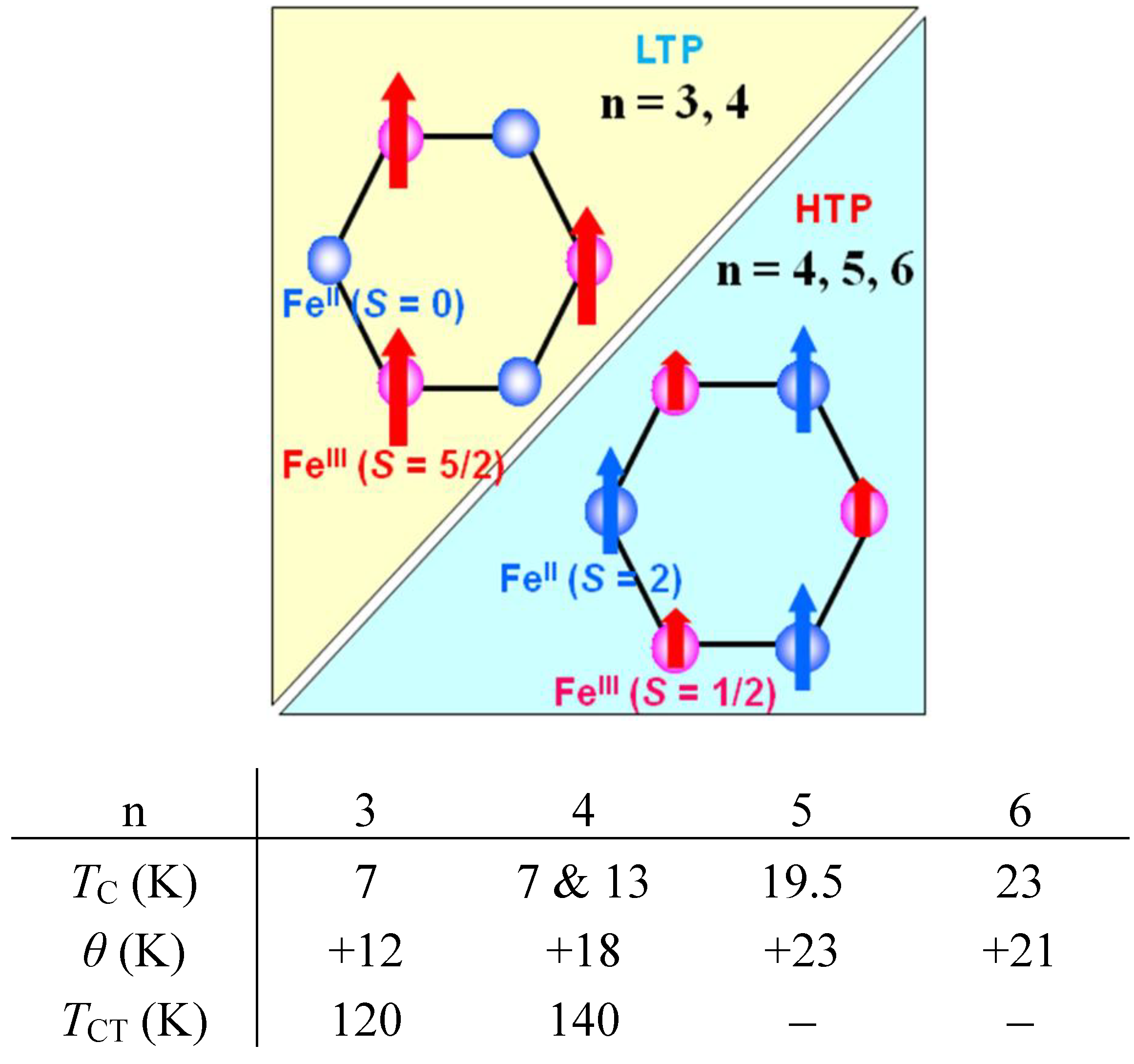
Figure 8.
Schematical feature of ferromagnetism and the magnetic parameters for (n-CnH2n+1)4N[FeIIFeIII(dto)3](n = 3–6). TCT denotes the critical temperature of the charge transfer phase transition.
Figure 8.
Schematical feature of ferromagnetism and the magnetic parameters for (n-CnH2n+1)4N[FeIIFeIII(dto)3](n = 3–6). TCT denotes the critical temperature of the charge transfer phase transition.
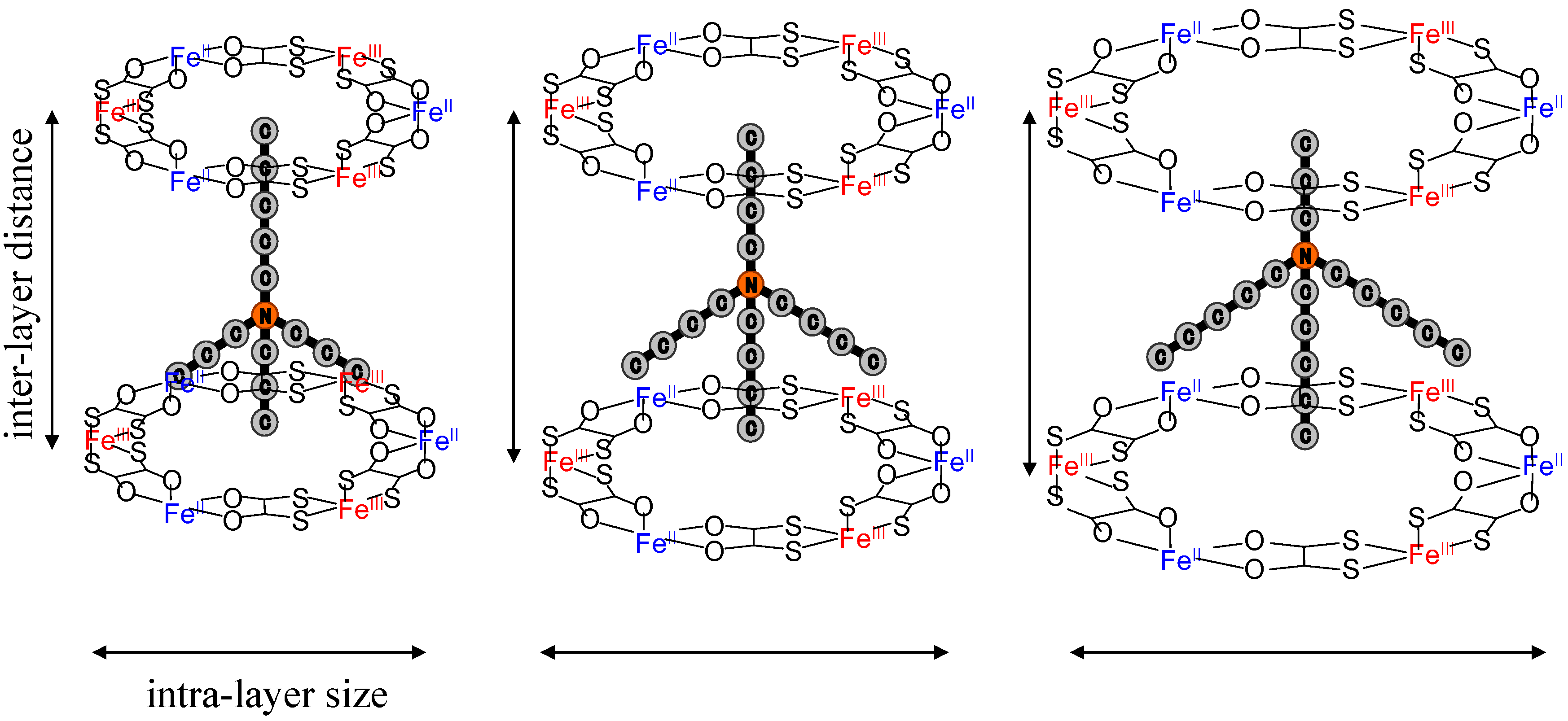
2.3. Cation size effect on the crystal structure
As mentioned in the previous section, the CTPT of (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3] exhibits a significant dependence on the size of counter cation. In order to investigate the more detailed structural factor that controls the CTPT, we have synthesized iron mixed valence complexes with various size of counter cations, (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3] [
61]. The series of symmetrical m = n system gives the expansion of both the inter- and intra-layer distances with increasing n value, while the uniaxial m ≠ n system would expand the inter- or intra-layer distance, independently, if the (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N cations are located at a three-fold axis and the alkyl chain of
n-C
nH
2n+1 points into the center of the hexagonal cavity surrounded by [Fe
IIFe
III(dto)
3] as shown in
Figure 9. Hereafter, (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3] as well as (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3] are denoted as (
m,
n); e.g., (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] is (3, 3). As for (m, n), (m, n) with
m +
n = 8 were investigated because (4, 4) shows the bistable state of the HTP and LTP below
TCT ~ 140 K as described hereinafter, so that the electronic state of Fe(II, III) is presumably sensitive to the structural change induced by substituting the counter cation. Since single crystals of (2, 6), (3, 5), (5, 3), (6, 2), (4, 4), (5, 5), and (6, 6) were not obtained, the structural characteristics of them were investigated by the powder X-ray diffraction (PXRD) at room temperature.
Figure 9.
Schematic representation of a part of two-dimensional network structure modified by axially symmetrical counter cations in (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3] (
m +
n = 8) [
61].
Figure 9.
Schematic representation of a part of two-dimensional network structure modified by axially symmetrical counter cations in (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3] (
m +
n = 8) [
61].
Comparing the powder pattern between (3, 3) and (4, 4), both of (3, 3) and (4, 4) have the space group of P63. In these complexes, the lattice parameters were obtained by Rietveld method. The lattice parameter, a, corresponds to the intra-layer distance of FeII-FeII (= FeIII-FeIII) and the lattice parameter, c/2, corresponds to the inter-layer distance (≡ d) of the [FeIIFeIII(dto)3] layer.
In the case of (5, 5), the PXRD profile cannot be refined in
P6
3, while it can be indexed using
P6
3 and trigonal
R3 space groups, which implies that (5, 5) is a biphasic complex. In connection with this, many bimetallic 2D oxalato complexes exhibiting trigonal
R3c space group have been reported [
27,
28,
29]. In the cases of polycrystalline A[M
IIFe
III(ox)
3] (A = (
n-C
3H
7)
4N, (
n-C
4H
9)
4N; M = Mn, Fe), on the other hand, the PXRD profiles show biphasic corresponding to the coexistence of
P6
3 and
R3c phases [
57,
62,
63]. In general, layered materials quite often show the structural disorder arising from the translation of rigid planes. If there are finite numbers of possible translational vectors between successive layers, (
hkl) dependent broadening of peaks in the powder diffraction pattern arises, which is known as stacking fault. The polycrystalline (5, 5) may be also the similar case where the stacking is randomly faulted between each type, while the
R3c phase would become a lower symmetrical group, trigonal
R3. Since the
R3c structure has a six-layer in a unit cell,
a also gives the intra-layer nearest Fe
II-Fe
II (= Fe
III-Fe
III) distance but
c / 6 corresponds to the inter-layer distance
d.
In the case of polycrystalline sample of (6, 6), the similar pattern as (5, 5) is observed. This result suggests the biphasic structure of P63 and R3, although only d is estimated from the (002) reflection at the lowest angle because of a lack of peaks available.
Figure 10.
Temperature dependence of the molar magnetic susceptibility,
χMT, of (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3](
m +
n = 8) on cooling and heating processes. The external magnetic field is 5000 Oe [
61].
Figure 10.
Temperature dependence of the molar magnetic susceptibility,
χMT, of (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3](
m +
n = 8) on cooling and heating processes. The external magnetic field is 5000 Oe [
61].
In the cases of (m, n) with m ≠ n, the PXRD profiles of (3, 5) and (5, 3) were successfully refined in P63. The PXRD profile of (6, 2) shows the similar pattern to those of (5, 5) and (6, 6), and is assigned to the biphasic structure of P63 and R3.
2.4. Cation size effect on the CTPT and magnetism
In the case of the uniaxial cation intercalated complexes, the effective magnetic moments at 300 K for (2,6), (3,5), (5,3) and (6,2) are 5.80, 5.64, 5.84, and 5.92 μ
B, respectively. These values are consistent with the value from the HTP with Fe
II(
S = 2) and Fe
III(
S = 1/2). The magnetic susceptibilities for (2,6), (3,5), (5,3) and (6,2) obey the Curie-Weiss law, χ
–1 = (
T-θ) /
T, in the range of 150-300 K. The Weiss constants of (2,6), (3,5), (5,3) and (6,2) are estimated at +24.1 K, +27.9 K, +24.4 K and +21.3 K, respectively. All the positive Weiss constants imply the ferromagnetic interaction between Fe
II and Fe
III. In the cases of (2,6), (3,5) and (5,3), a small bump with thermal hysteresis loop appears in the χ
T curve around 100 K, which is shown in
Figure 10. This feature is quite similar to the CTPT for (3, 3) and (4, 4), which implies that the CTPT occurs in (2,6), (3,5) and (5,3). On the other hand, in the case of (6,2), the CTPT does not take place and HTP is stable even at 2 K.
Figure 11 shows the FCM, RM and ZFCM for (2,6), (3,5), (5,3) and (6,2). In the cases of (2,6), (3,5) and (5,3), reflecting the coexistence of LTP and HTP even at 2 K, the LTP and HTP undergo the ferromagnetic transitions individually, which induces the stepwise ferromagnetic transition. The Curie temperatures for (2,6), (3,5), (5,3) and (6,2) are 6.5 and 18 K, 5.5 and 17 K, 6.5 and 18 K and 17 K, respectively.
Figure 11.
Temperature dependences of FCM, RM, and ZFCM of (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3](
m +
n = 8). The external magnetic field is 30 Oe [
61].
Figure 11.
Temperature dependences of FCM, RM, and ZFCM of (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3](
m +
n = 8). The external magnetic field is 30 Oe [
61].
The spin states of (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3] with uniaxial cation are also confirmed by the
57Fe Mössbauer spectroscopy [
61]. The
57Fe Mössbauer spectra of (2,6), (3,5), (5,3) and (6,2) at 300 K show the similar patterns to those of (3, 3), (4,4), (5,5) and (6, 6) where two major quadrupole doublets attributed to the Fe
II (
S = 2) and Fe
III (
S = 1/2) sites. In the case of (6, 2), the spectrum is almost unchanged between 200 and 77 K. On the other hand, in the cases of (2, 6), (3,5) and (5, 3), the spectra for Fe
II(
S = 2) and Fe
III(
S = 1/2) decrease and new peaks corresponding to the superposition of Fe
II (
S = 0) and Fe
III (
S = 5/2) appear at 77 K. This feature is quite similar to the
57Fe Mössbauer spectra of (3, 3) and (4, 4) at 77 K.
From the results of the magnetic measurements and the
57Fe Mössbauer spectroscopy, the following feature is concluded. In the cases of (2, 6), (3,5) and(5, 3), as well as (4, 4), the CTPT occurs at around 100 K but the transition is incomplete so that the HTP and LTP coexist even at low temperature region, and consequently the individual ferromagnetic phase transitions due to the HTP and LTP are observed. On the other hand, the HTP remains in all the measuring temperature range for (6, 2) as well as the spin states for (5, 5) and (6, 6).
Figure 12 shows the relationship between the spin states and the intercalated cations for (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
3] at 77 K, where the white and black colored areas correspond to the LTP and HTP, respectively.
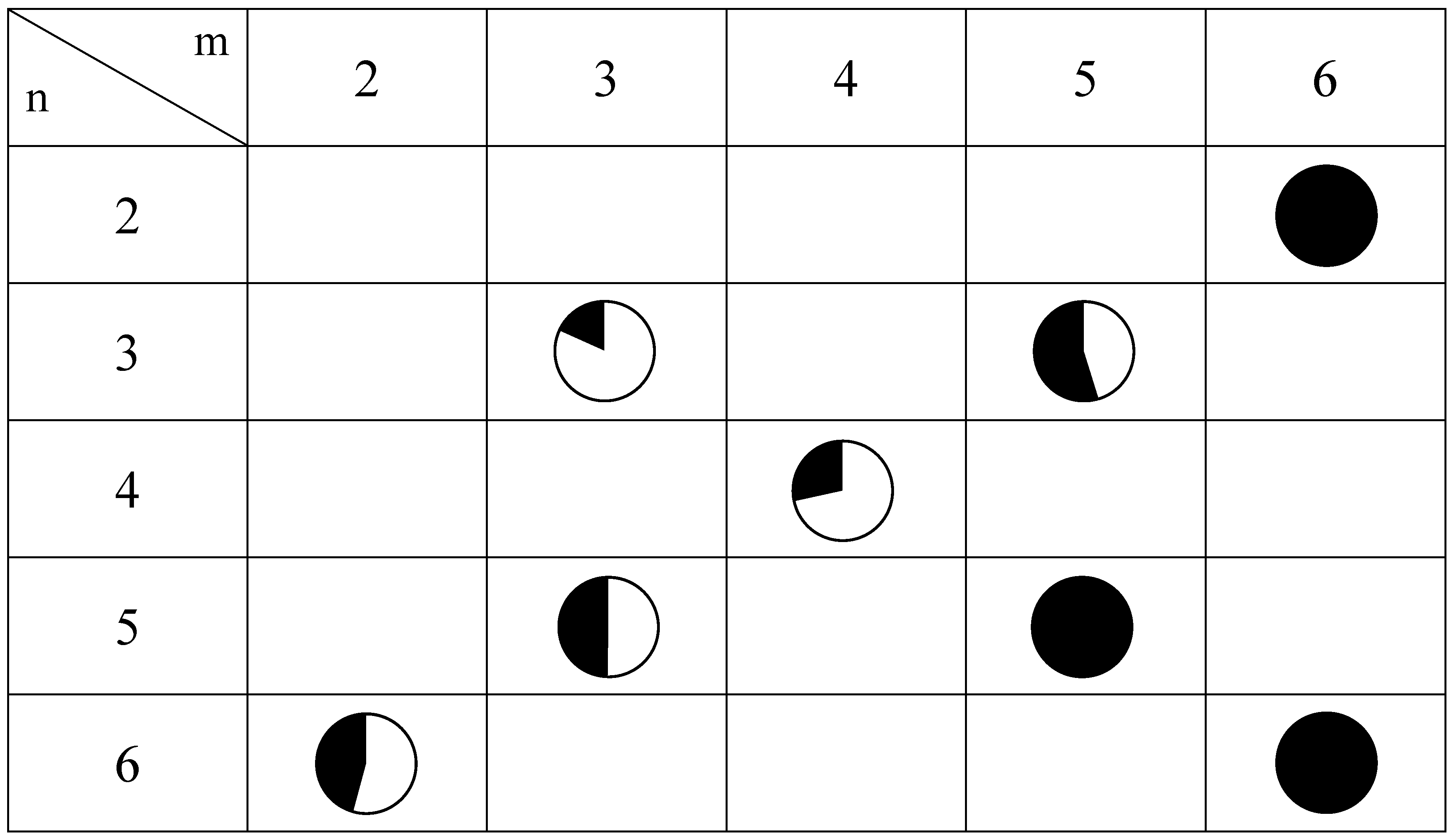
Figure 12.
Mixing ratio between LTP and HTP for (n-CmH2m+1)3(n-CnH2n+1)N[FeIIFeIII(dto)3] obtained by the 57Fe Mössbauer spectroscopy at 77 K. White and black areas correspond to the LTP and HTP fractions, respectively.
Figure 12.
Mixing ratio between LTP and HTP for (n-CmH2m+1)3(n-CnH2n+1)N[FeIIFeIII(dto)3] obtained by the 57Fe Mössbauer spectroscopy at 77 K. White and black areas correspond to the LTP and HTP fractions, respectively.
As shown in
Figure 12, the CTPT for the (
n-C
mH
2m+1)
3(
n-C
nH
2n+1)N[Fe
IIFe
III(dto)
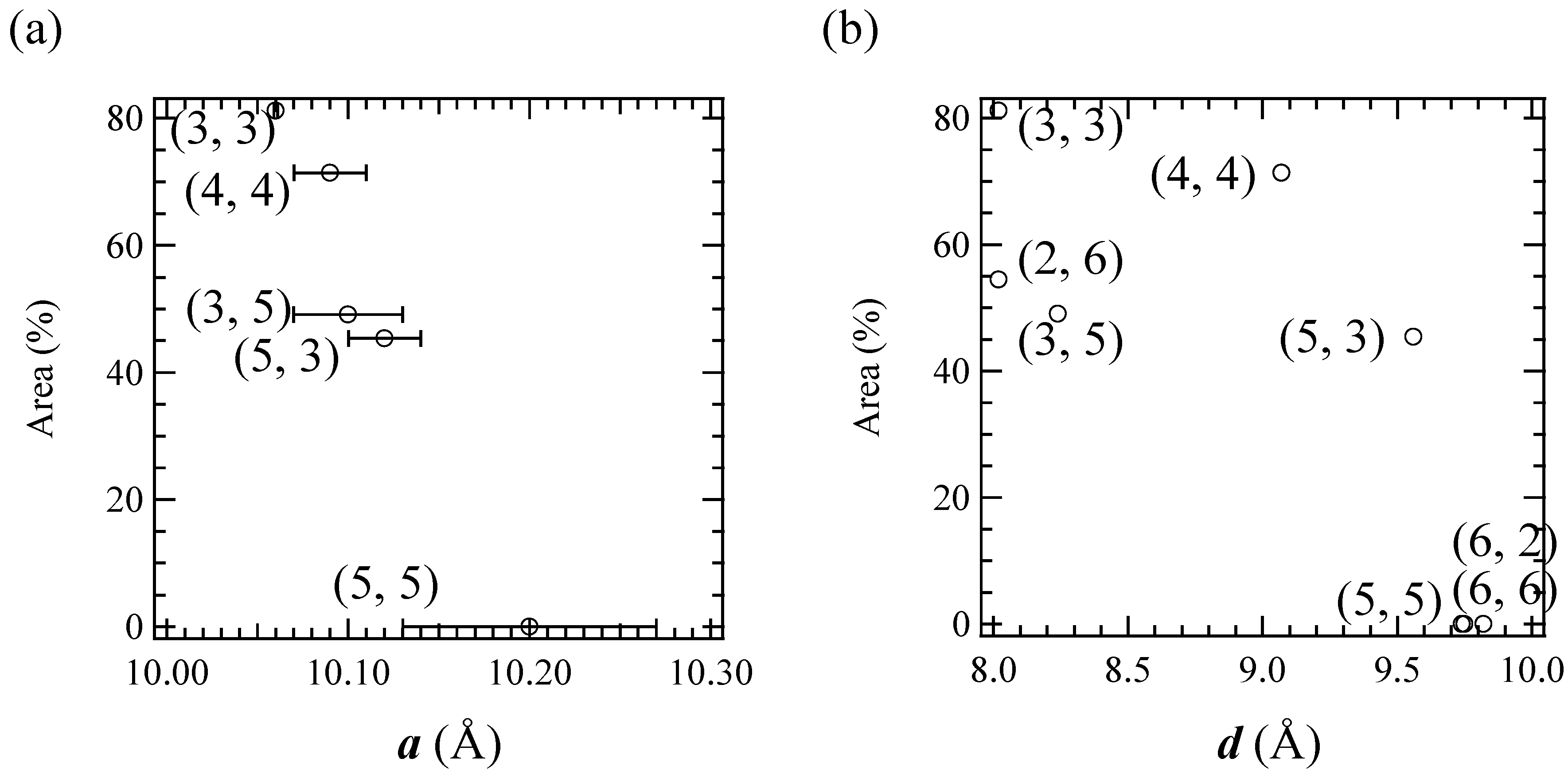
3] significantly exhibits a cation-size dependence. In order to clarify the correlation between the stability of the LTP and the crystal structure in this system, the relationship between the unit cell parameters and the area of the
57Fe Mössbauer spectra corresponding to the LTP at 77 K are plotted, which is shown in
Figure 13. The correlation between the cell parameter
a and the fraction of LTP at 77 K is shown in
Figure 13(a). The fraction of the LTP tends to decrease with increasing
a, then becomes zero at
a = 10.2±0.07 Å. On the other hand, the correlation between the cell parameter
d and the fraction of LTP at 77 K is not clear. Therefore it can be concluded that the bulkier cation expands the intra-layer nearest Fe
II-Fe
II (= Fe
III-Fe
III) distance in the 2D honeycomb network structure of [Fe
IIFe
III(dto)
3], which directly stabilizes the HTP and hence the CTPT is suppressed in the system.
Figure 13.
The cell parameter (a) a and (b) d dependences of the area of the 57Fe Mössbauer spectrum corresponding to the LTP at 77 K. In (a), selected (n-CmH2m+1)3(n-CnH2n+1)N[FeIIFeIII(dto)3] complexes whose a were obtained are plotted.
Figure 13.
The cell parameter (a) a and (b) d dependences of the area of the 57Fe Mössbauer spectrum corresponding to the LTP at 77 K. In (a), selected (n-CmH2m+1)3(n-CnH2n+1)N[FeIIFeIII(dto)3] complexes whose a were obtained are plotted.
2.5. Valence fluctuation at the CTPT
As mentioned in the previous section, the charge transfer phase transition (CTPT) occurs in (n-C3H7)4N[FeIIFeIII(dto)3] (hereafter denoted as n = 3), while (n-C5H11)4N[FeIIFeIII(dto)3] (likewise, n = 5) keeps its spin state as the HTP in the whole measuring temperature range. Although the understanding of the mechanism of electron transfer in such materials is of great importance, the nature of the CTPT, dynamic properties of electron transfer in particular, has not been fully understood. The most important interest about the CTPT is whether electrons are statically localized or dynamically fluctuated between the FeII and FeIII sites under thermal equilibrium condition.
To reveal the dynamics of the CTPT, muon spin relaxation (μSR) is the most powerful technique. Since the μSR is based on the observation of the evolution with time of the direction of the muon spin in the magnetic field at the muon site in the sample, it is a very useful technique that probes the magnitude, distribution, and dynamics of the internal fields. Moreover, since μSR has a wider characteristic time window (typically from 10−5 to 10−11 sec) than the 57Fe Mössbauer spectroscopy (around 10−7 sec), μSR can be a more sensitive microscopic probe to sense the dynamics of CTPT within a wider frequency range rather than 57Fe Mössbauer spectroscopy.
In this section, the results of zero-field (ZF) and longitudinal-field (LF) μSR measurements for n = 3 and 5 are reported to investigate the dynamics of the electron transfer between FeII and FeIII, which is accompanied by the CTPT.
2.5.1. Zero-field muon spin relaxation
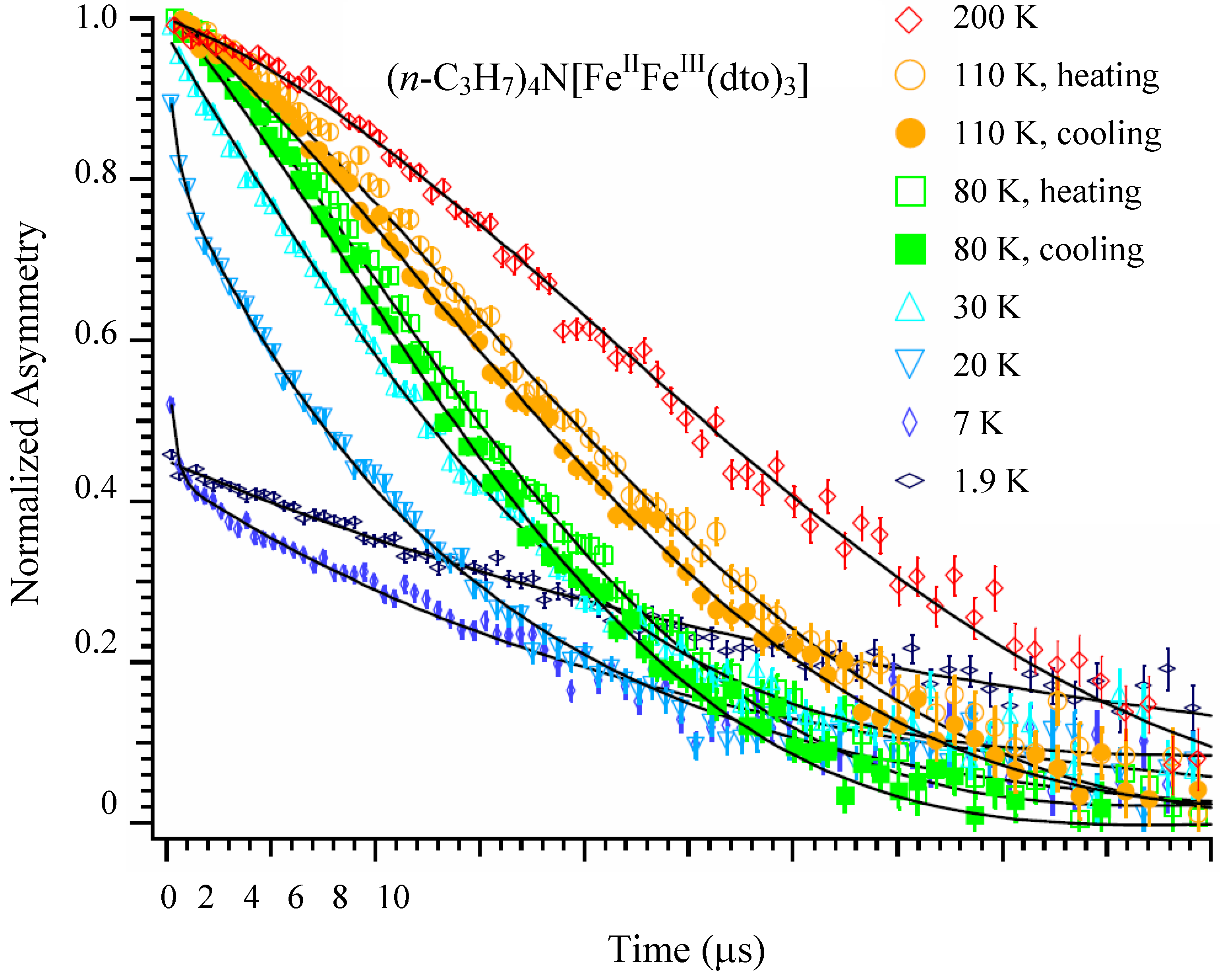
Figure 14 shows the ZF-μSR time spectra of n = 3 between 1.9 and 200 K [
41]. A Gaussian-type depolarization behavior of the time spectrum is observed at 200 K. The time spectrum changes to an exponential type with decreasing temperature below 30 K, and a loss of the initial asymmetry is observed below 20 K. Within a time region longer than 1 μs, the time spectrum slightly recovers with decreasing temperature down to 1.9 K. This series of behaviors is a sign of the appearance of the ferromagnetically ordered state (
TC = 7 K). Furthermore, thermal hysteresis is observed around 120 K, at which the CTPT occurs. The time spectra obtained at 80 and 110 K in the cooling process are always located below those obtained at the same temperatures in the heating process. This means that the muon spin depolarizes faster in the cooling process than in the heating process. The tendency of hysteresis in the temperature dependence of the muon spin depolarization behavior around 80 K is consistent with that observed in our previous magnetic susceptibility measurement [
32].
Figure 14.
Zero-field μSR time spectra of (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3]. The solid lines show the best fit of fitting function. The spectra at 80 and 110 K show both the heating and the cooling processes [
41].
Figure 14.
Zero-field μSR time spectra of (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3]. The solid lines show the best fit of fitting function. The spectra at 80 and 110 K show both the heating and the cooling processes [
41].
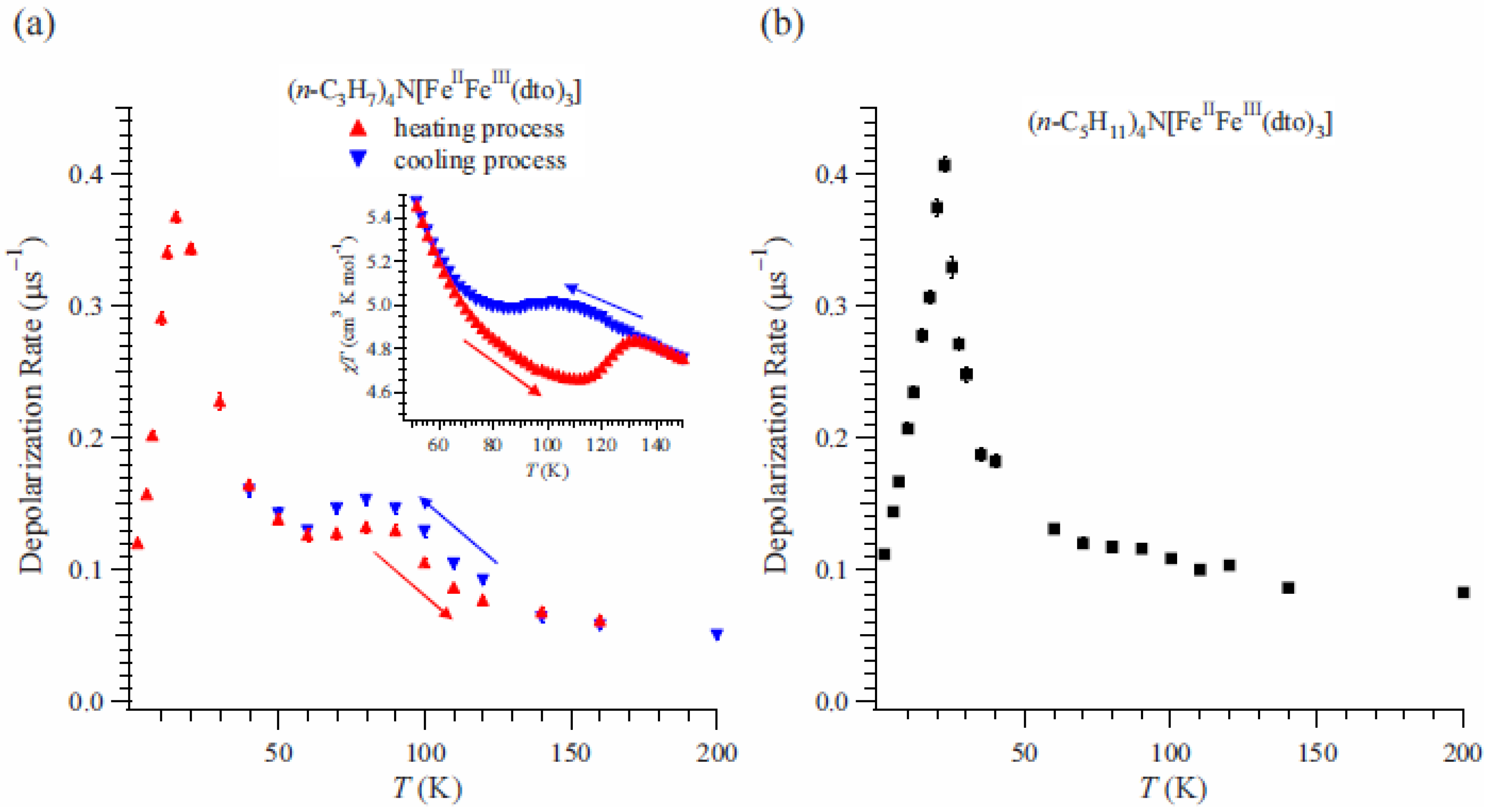
The depolarization rate, λ
0, of n = 3 can be obtained by fitting of these time spectra as shown in
Figure 15(a) [
41]. λ
0 exhibits a peak at 15 K. Since the ferromagnetic transition temperature has been estimated at 7 K from the susceptibility measurement, the enhancement of λ
0 above 15 K is due to the critical slowing down of the fluctuations of Fe spins toward the ferromagnetic transition [
64]. Moreover, an anomalous enhancement with thermal hysteresis of λ
0 was observed between 60 and 140 K. The thermal hysteresis of λ
0 is presumably caused by the changes in the dynamic properties of electrons. The range of temperatures wherein the anomalous enhancement of λ
0 was observed is well matched with the temperature range wherein the CTPT appears (see the inset of
Figure 6(a)).
Figure 15.
(
a) Temperature dependence of the dynamical muon spin depolarization rate, λ
0, for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] on the cooling and heating processes. The inset shows the temperature dependence of the magnetic susceptibility for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3]. ← and → denote the heating and cooling processes, respectively. (
b) Temperature dependences of λ
0 for (
n-C
5H
11)
4N[Fe
IIFe
III(dto)
3] [
41].
Figure 15.
(
a) Temperature dependence of the dynamical muon spin depolarization rate, λ
0, for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] on the cooling and heating processes. The inset shows the temperature dependence of the magnetic susceptibility for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3]. ← and → denote the heating and cooling processes, respectively. (
b) Temperature dependences of λ
0 for (
n-C
5H
11)
4N[Fe
IIFe
III(dto)
3] [
41].
We also measured the ZF-μSR of n = 5 for the comparison because both the thermal hysteresis of the susceptibility and the CTPT were not observed [
40]. The time spectra for n = 5 were analyzed in the same way as for n = 3.
Figure 15(b) displays the temperature dependence of λ
0 for n = 5 [
41], where λ
0 increases with decreasing temperature below 200 K. Values of λ
0 for n = 5 are similar to those for n = 3 at the measuring temperatures except for the temperature region wherein the CTPT occurs. This fact means that depolarization mechanism of the muon spin in the case of n = 5 is similar to that of n = 3 except for the effect of CTPT. A strong enhancement was observed around 22 K, which is due to the critical slowing down of Fe spins toward the ferromagnetically ordered state like the case of n = 3. Since the anomalous enhancement of λ
0 with thermal hysteresis around 80 K is observed only for n = 3, it is concluded that it originates from the CTPT. Taking into account that the CTPT is accompanied by the electron transfer and the HTP state is mixed with the LTP state around 80 K in the case of n = 3, it can be concluded that the motion of electrons between the Fe
II and Fe
III sites induces fluctuating internal fields at the muon site enhancing λ
0. Therefore, the oscillation of electrons between the Fe
II and Fe
III sites is the intrinsic nature of CTPT.
2.5.2. Longitudinal-field muon spin relaxation
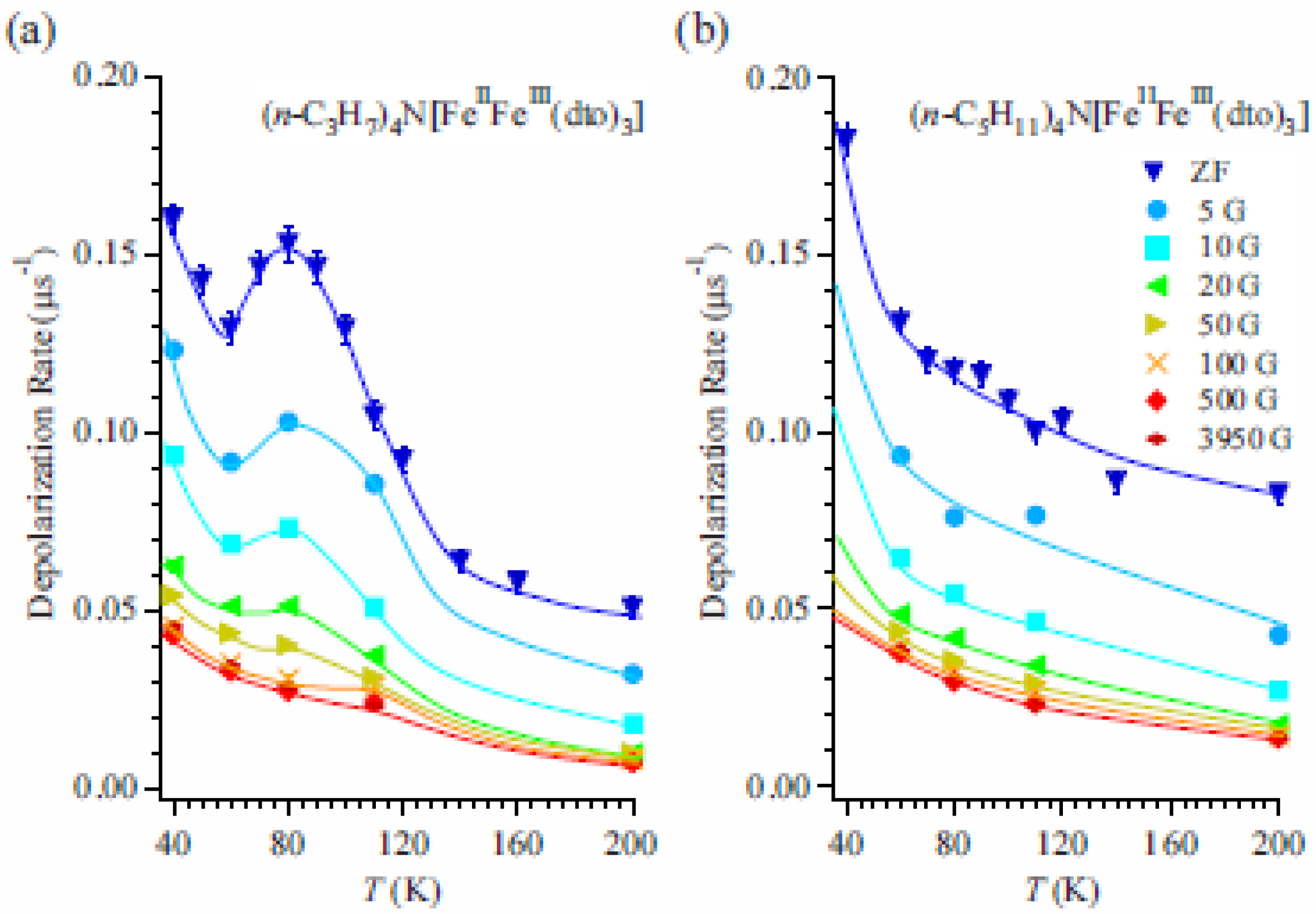
To obtain more detailed information on the dynamical properties of the CTPT, LF-μSR was performed at 80 K in the cooling process for both n = 3 and 5.
Figures 16(a) and (b) display the temperature and LF dependences of λ
0 for n=3 and 5, respectively. In the case of n = 3, the peak around 80 K disappears with increasing LF. λ
0 for both n = 3 and 5 decreases with increasing LF up to about 100 Oe and becomes constant above 100 Oe. The constant values increase with decreasing temperature. These results suggest the existence of two components in the LF dependence of λ
0. One is easily suppressed by a weak LF of about 100 Oe and shows a convex shape in its LF dependence (the weak component), and the other is independent of the LF up to 4 kOe (the strong component). The weak component means that there is a small and slowly fluctuating internal field at the muon site, which is easily masked by a small LF. Although the origin of the weak component is not clear at the moment, it would be suggested to be due to the fluctuating component of nuclear dipoles as was observed in MnSi [
65]. As for the strong component, considering that the value of the strong component increases with decreasing temperature and similar values of λ
0 are observed in both cases of n = 3 and 5 at the same temperature, it is suggested that the strong component originates from the dipole field of dynamically fluctuating Fe spins. Then, the electron transfer due to the CTPT has an additional effect on the weak component of the muon spin depolarization. This means that electrons oscillating between the Fe
II and Fe
III sites make dynamically fluctuating internal fields at the muon site.
Figure 16.
(
a) Temperature and longitudinal-field dependence of the dynamic muon spin depolarization rate, λ
0, for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] and (
b) (
n-C
5H
11)
4N[Fe
IIFe
III(dto)
3] [
41]. The solid lines are guides for the eye.
Figure 16.
(
a) Temperature and longitudinal-field dependence of the dynamic muon spin depolarization rate, λ
0, for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] and (
b) (
n-C
5H
11)
4N[Fe
IIFe
III(dto)
3] [
41]. The solid lines are guides for the eye.
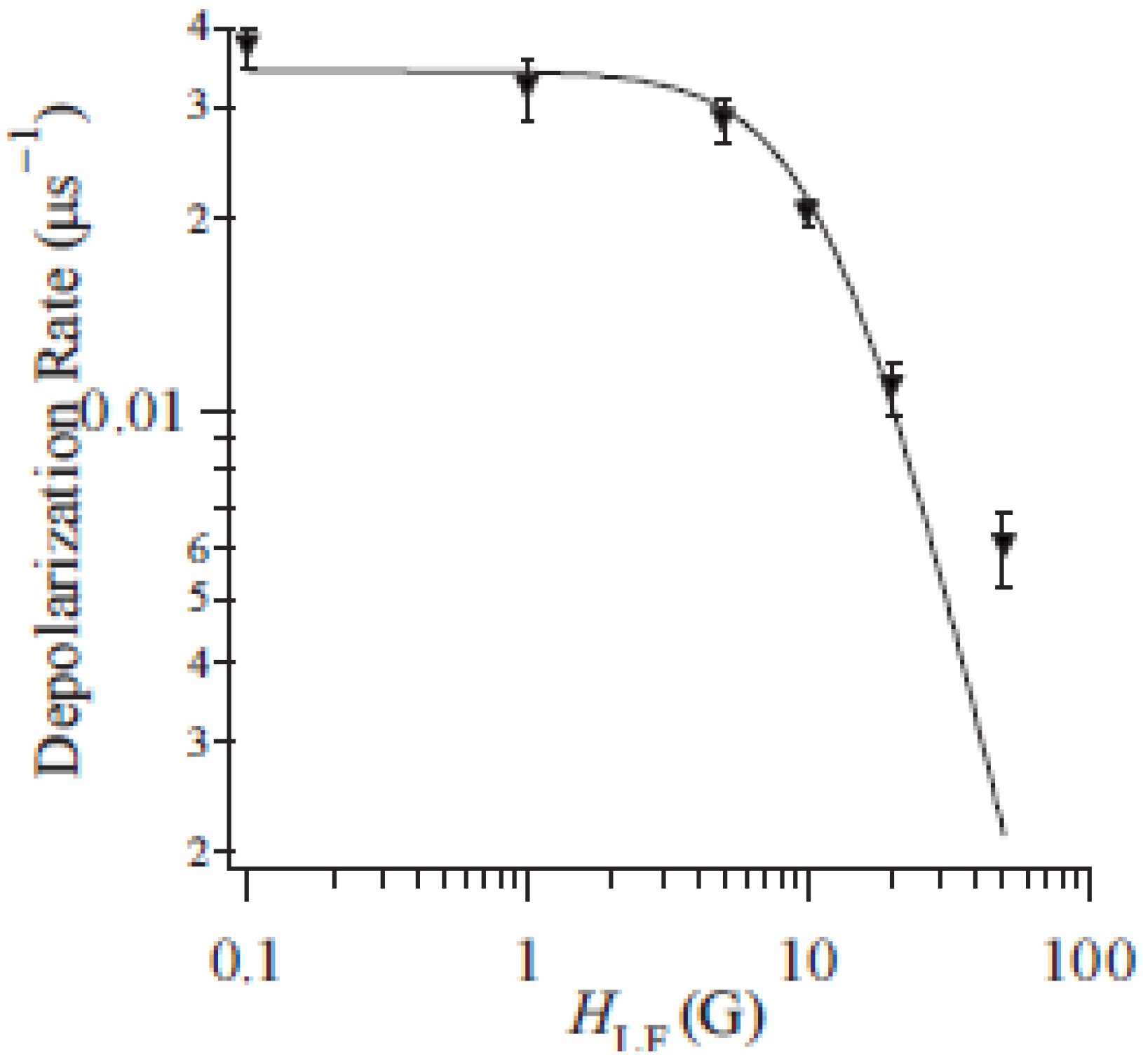
The weak component can be extracted from the difference of depolarization rate between n = 3 and 5. The LF dependence of this subtracted depolarization rate λ
CT at 80 K is summarized in
Figure 17. The LF dependence of λ
CT shows a convex shape as a function of
HLF in a log-log plot and tends to disappear around 100 Oe. This behavior depends on the correlation time of muon spins, τ
c, and the amplitude of the fluctuating internal field,
Hloc, at the muon site, respectively. τ
c is given as 5.7 μs from the analysis of the LF dependence of λ
CT. The frequency of the additional internal field at the muon site, which is given as
v = 1 / τ
c, is on the order of 0.1 MHz at 80 K. Moreover,
Hloc is estimated at 4.0 Oe, which is larger than that of contribution of the nuclear-dipole field (~1 Oe).
Figure 17.
Longitudinal-field dependence of the dynamical muon spin depolarization rate, λ
CT, for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at 80 K in the cooling process. The solid line shows the best fit of fitting function [
41].
Figure 17.
Longitudinal-field dependence of the dynamical muon spin depolarization rate, λ
CT, for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at 80 K in the cooling process. The solid line shows the best fit of fitting function [
41].
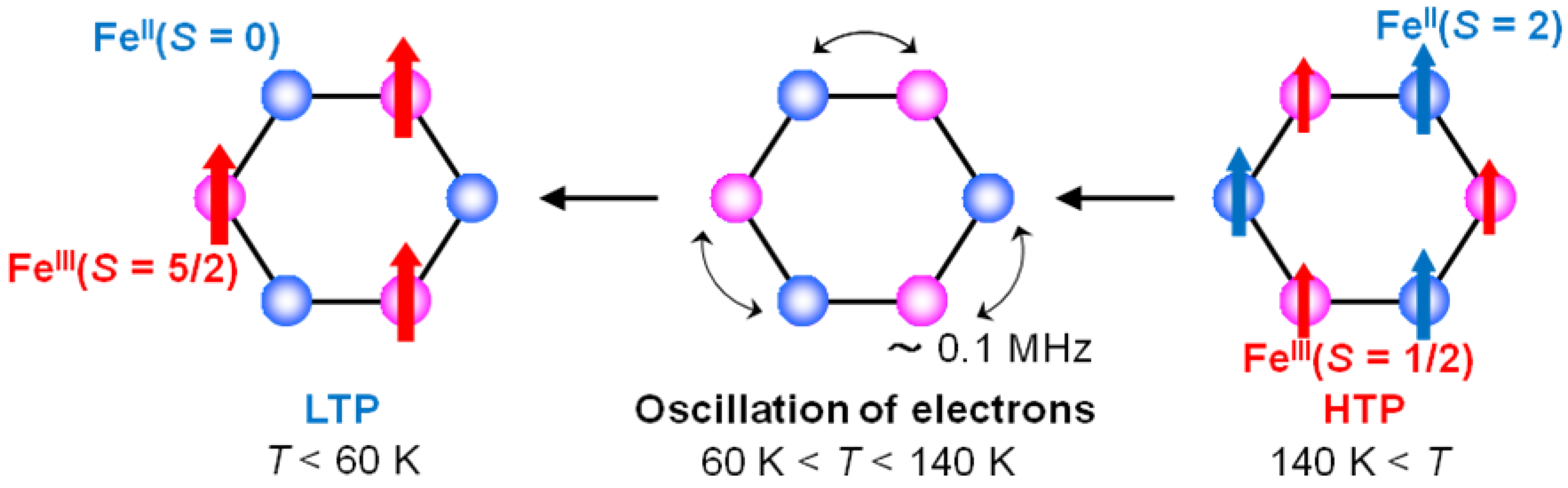
In the temperature range corresponding to the CTPT, it can be suggested that the flip flop of a moving electron between Fe
II and Fe
III sites produces the dynamically fluctuating internal field at the muon site and its frequency is represented by
v, which is schematically shown in
Figure 18. The time scale of τ
c is consistent with the result of the
57Fe Mössbauer measurement, which implies that the fluctuation between the HTP and the LTP is slower than 10
−7 s.
Figure 18.
The schematic feature of the electronic oscillation in the CTPT region for (n-C3H7)4N[FeIIFeIII(dto)3].
Figure 18.
The schematic feature of the electronic oscillation in the CTPT region for (n-C3H7)4N[FeIIFeIII(dto)3].
It was reported that the resistivity of n = 3 shows an anomalous drop with the thermal hysteresis loop within the temperature range of the CTPT [
66]. Considering the present μSR results, it can be suggested that the hopping conduction in the [Fe
IIFe
III(dto)
3] network is induced by the CTPT.
This result is the first observation of the dynamic electron transfer process of such mixed-valence complexes by using the μSR technique. Therefore, μSR will open the initiating research on the dynamics of charge-transfer phenomena for various inorganic and organic charge-transfer complexes that exhibit CTPT or neutral-ionic phase transitions.
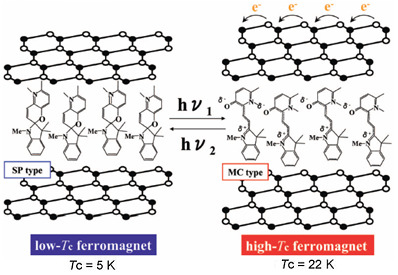
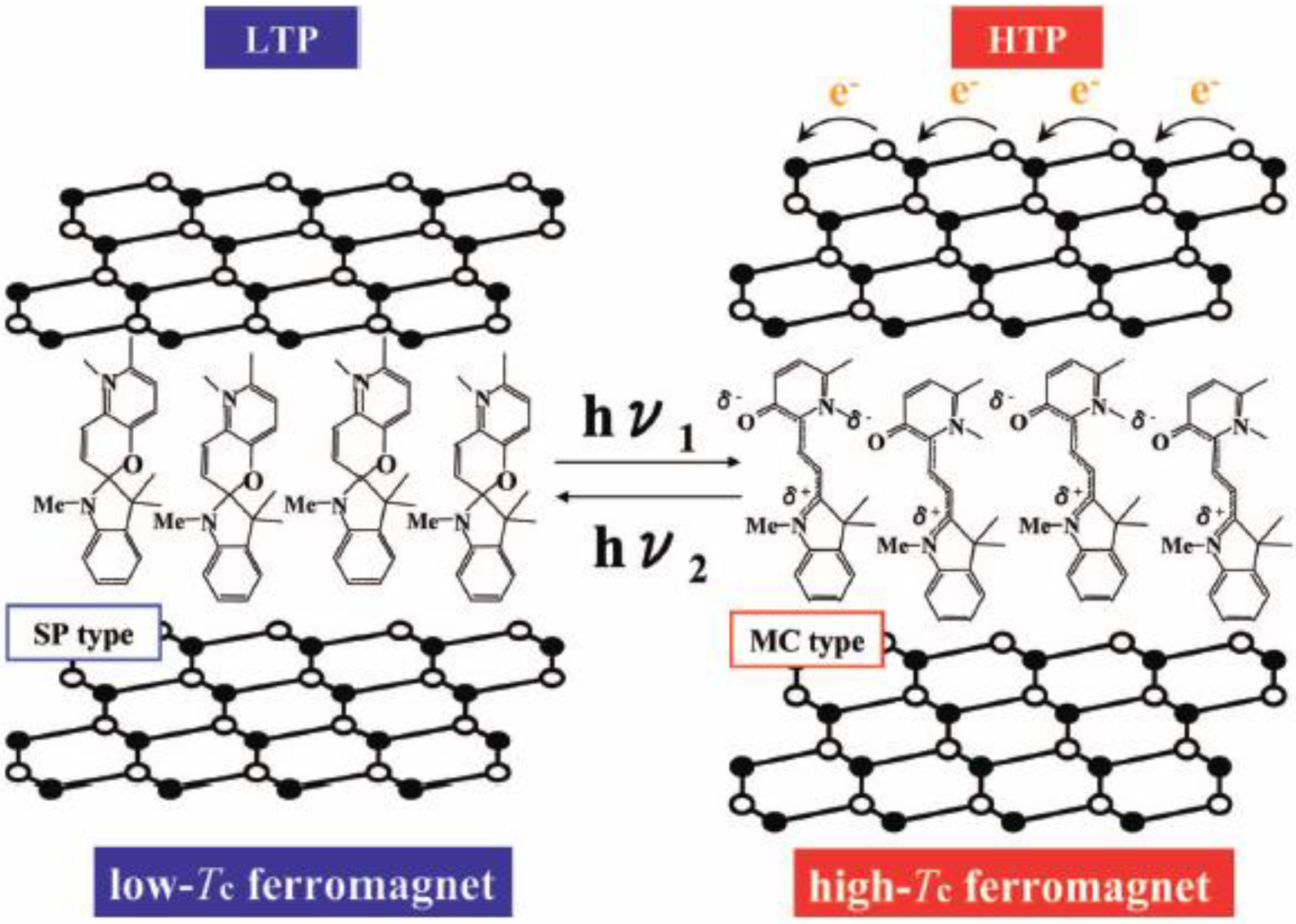
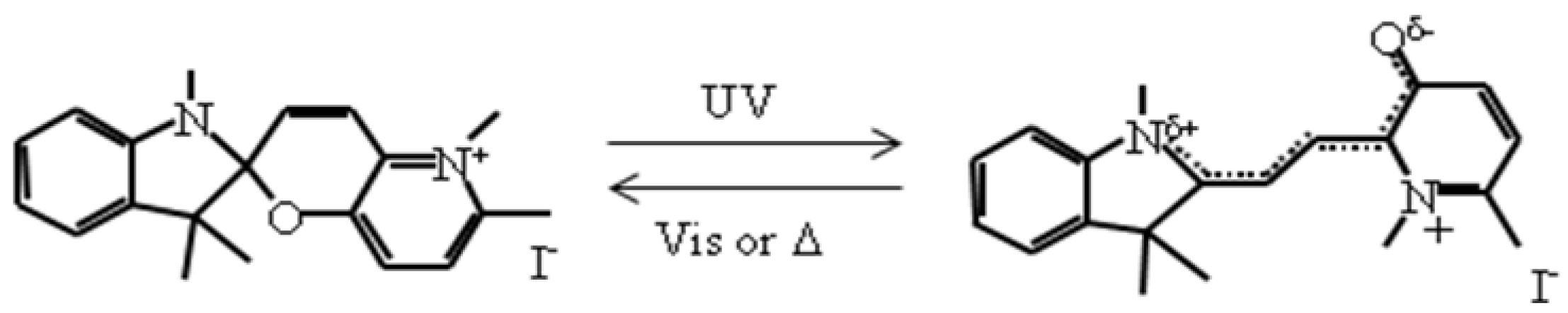
2.6. Concerted phenomenon coupled with photoisomerization and CTPT
As mentioned in section 2.3 and 2.4, the existence of the CTPT and the ferromagnetic transition in the [Fe
IIFe
III(dto)
3] system strongly depends on the size of intercalated cation. If the volume of the intercalated cation can be controlled by external stimuli, the CTPT becomes a controllable phenomenon. In organic-inorganic hybrid systems, it is effective to use an organic photochromic molecule for producing photoswitchable materials. On the basis of this strategy, we have used a photochromic spiropyran (SP) as the intercalated cation for [Fe
IIFe
III(dto)
3] and have tried to control the CTPT and the ferromagnetism for (SP)[Fe
IIFe
III(dto)
3] by means of photoisomerization of SP [
42]. In general, the cationic spiropyran is converted from the yellow-colored closed form (CF) to the red-colored open form (OF) upon the irradiation of UV light (330-370 nm) at room temperature. The OF is usually less stable and returns to the closed form both thermally and photochemically (500-600 nm) in solution. This photoisomerization is associated with the large volume change. In the case of several kinds of spiropyran derivative, photoisomerization occurs in the solid state [
67].
In this section, the photoinduced effect on the CTPT and the ferromagnetic transition of photosensitive organic-inorganic hybrid complexes, (SP-R)[Fe
IIFe
III(dto)
3] (SP-R = cationic spiropyran shown in
Scheme 1, R = methyl (Me), propyl (Pr)) are introduced.
Scheme 1.
Molecular structure of spiropyran.
Scheme 1.
Molecular structure of spiropyran.
2.6.1.Crystal structure
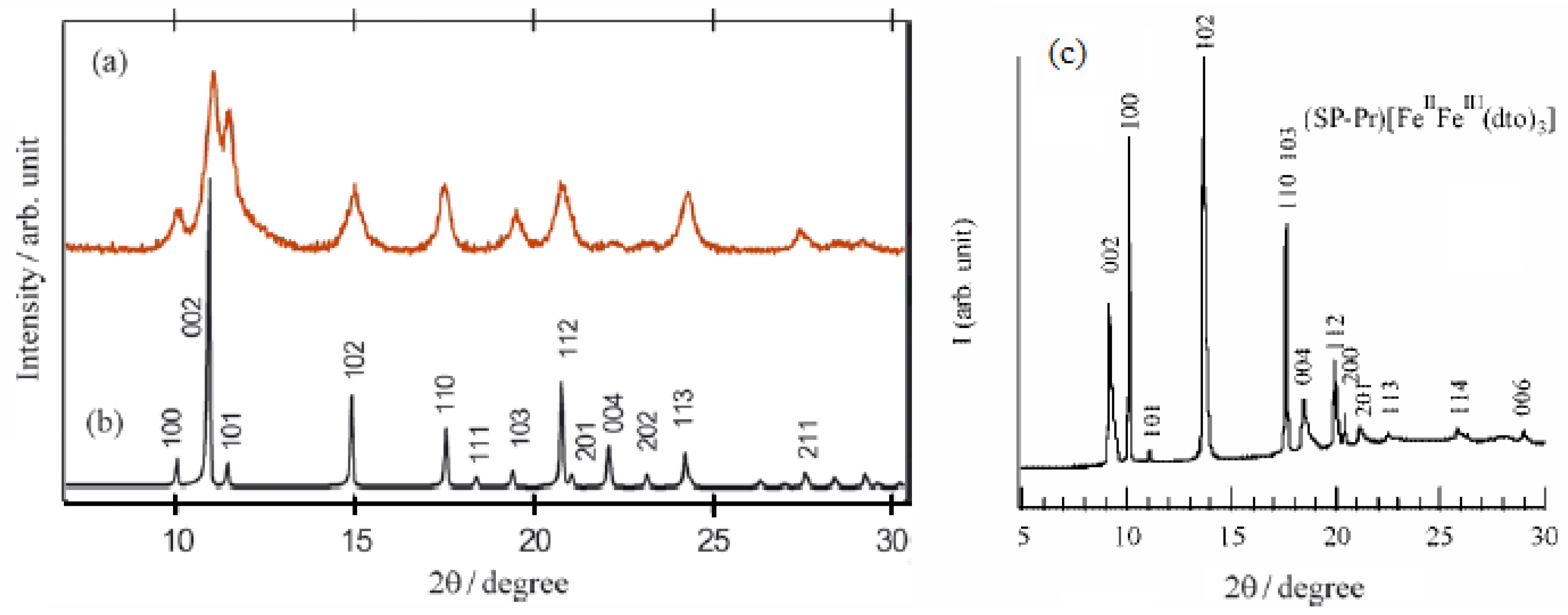
The powder X-ray diffraction patterns for (SP-Pr)[Fe
IIFe
III(dto)
3] (denoted as SP-Pr complex) and (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] are shown in
Figure 19 [
42]. The reflection for SP-Pr complex is indexed using a hexagonal unit cell just like most dto-bridged bimetal compounds with 2D honeycomb network structure, A[M
IIM’
III(dto)
3] (A = (n-C
nH
2n+1)
4N, (n-C
nH
2n+1)
4P, M = Mn, Co, Ni, Fe, M’ = Cr, Fe). The precise crystal structure of SP-Pr complex is isostructural with (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] since the peaks over the range 10°-22° suggest a hexagonal unit cell.
Figure 19.
Powder X-ray diffraction profiles of (
a) experimental diffraction profile for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at 300 K, (
b) calculated one on the space group of
P6
3 at 300 K, and (
c) experimental PXRD profile for (SP-Pr)[Fe
IIFe
III(dto)
3] [
42].
Figure 19.
Powder X-ray diffraction profiles of (
a) experimental diffraction profile for (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at 300 K, (
b) calculated one on the space group of
P6
3 at 300 K, and (
c) experimental PXRD profile for (SP-Pr)[Fe
IIFe
III(dto)
3] [
42].
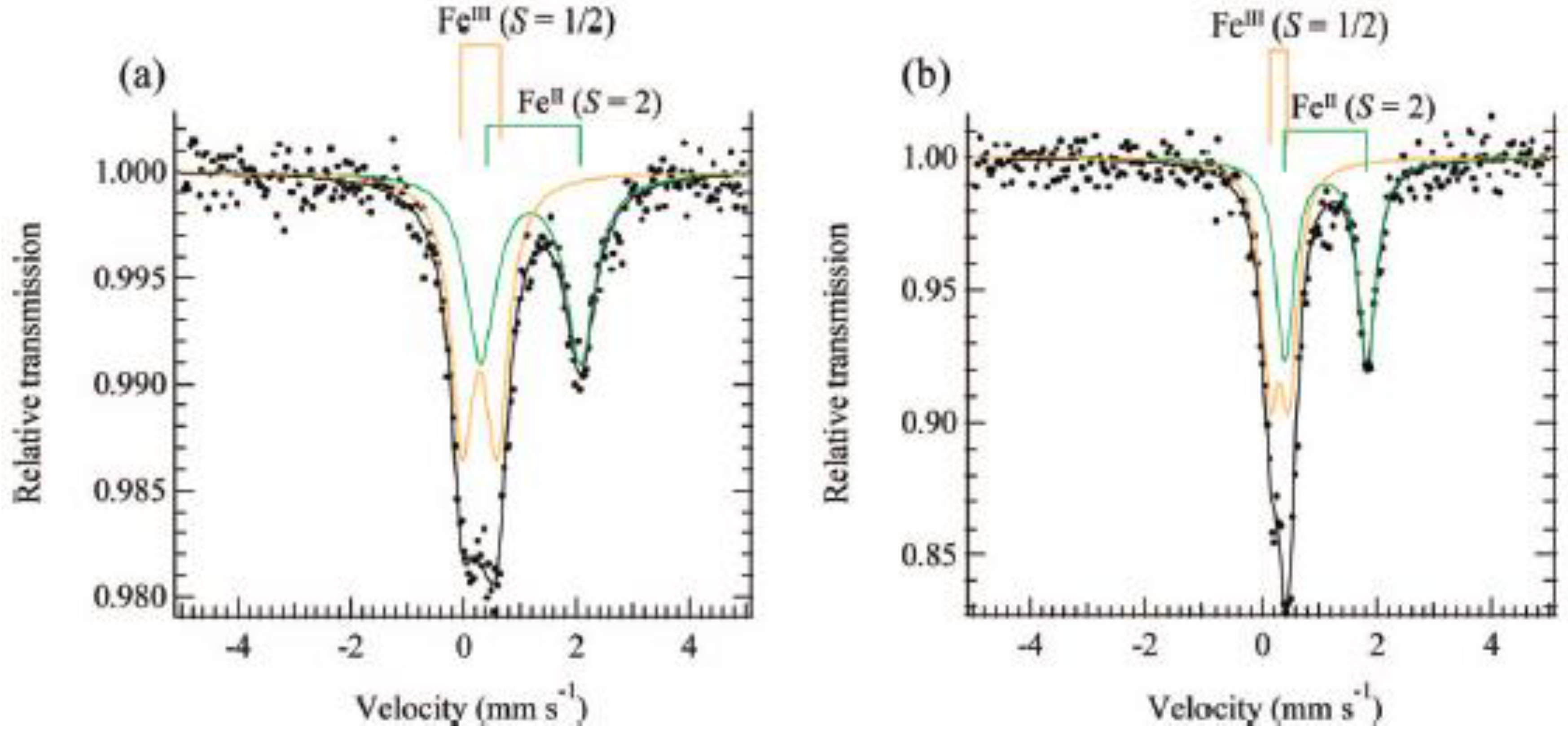
The
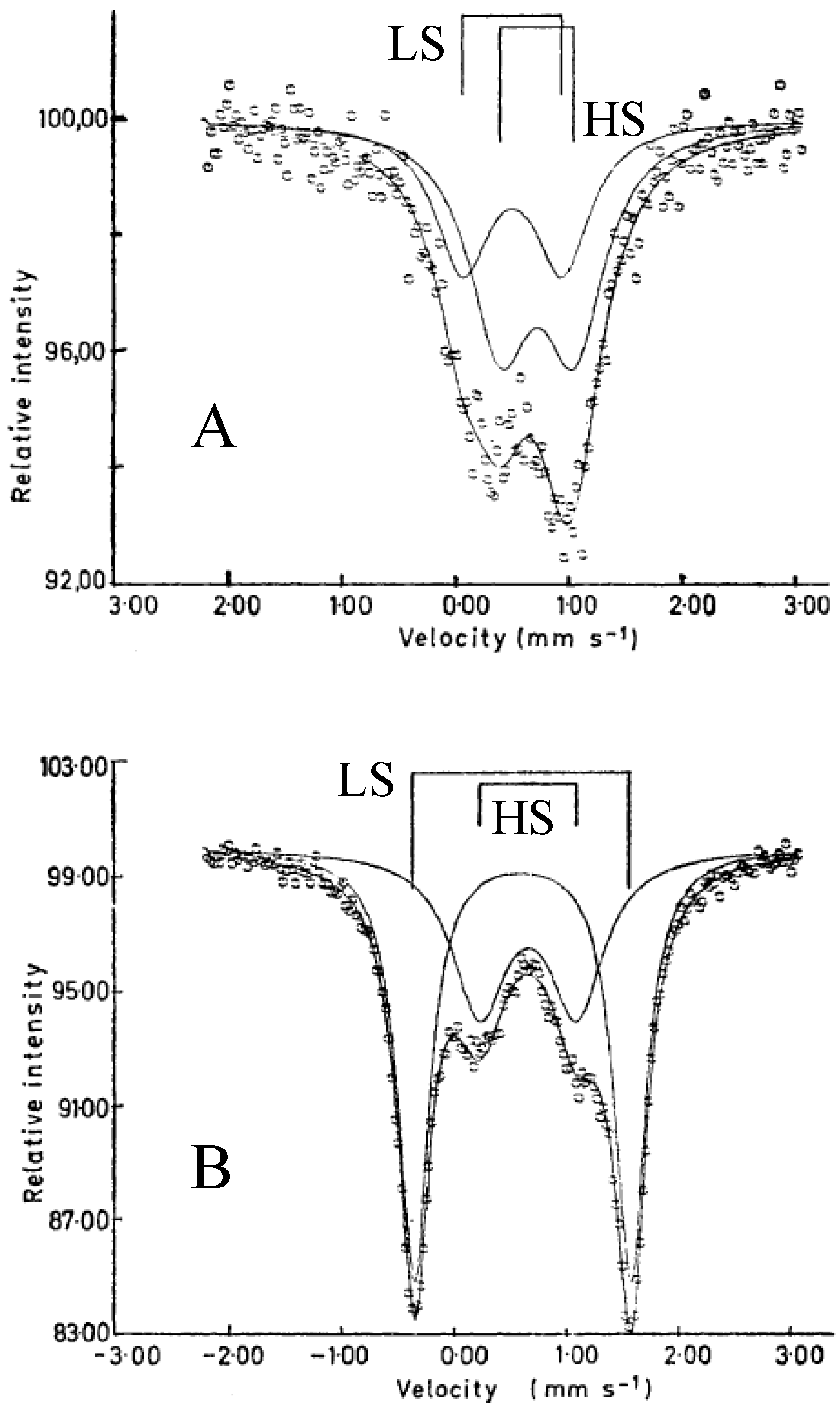
57Fe Mössbauer spectra of SP-Me complex and (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at room temperature are shown in
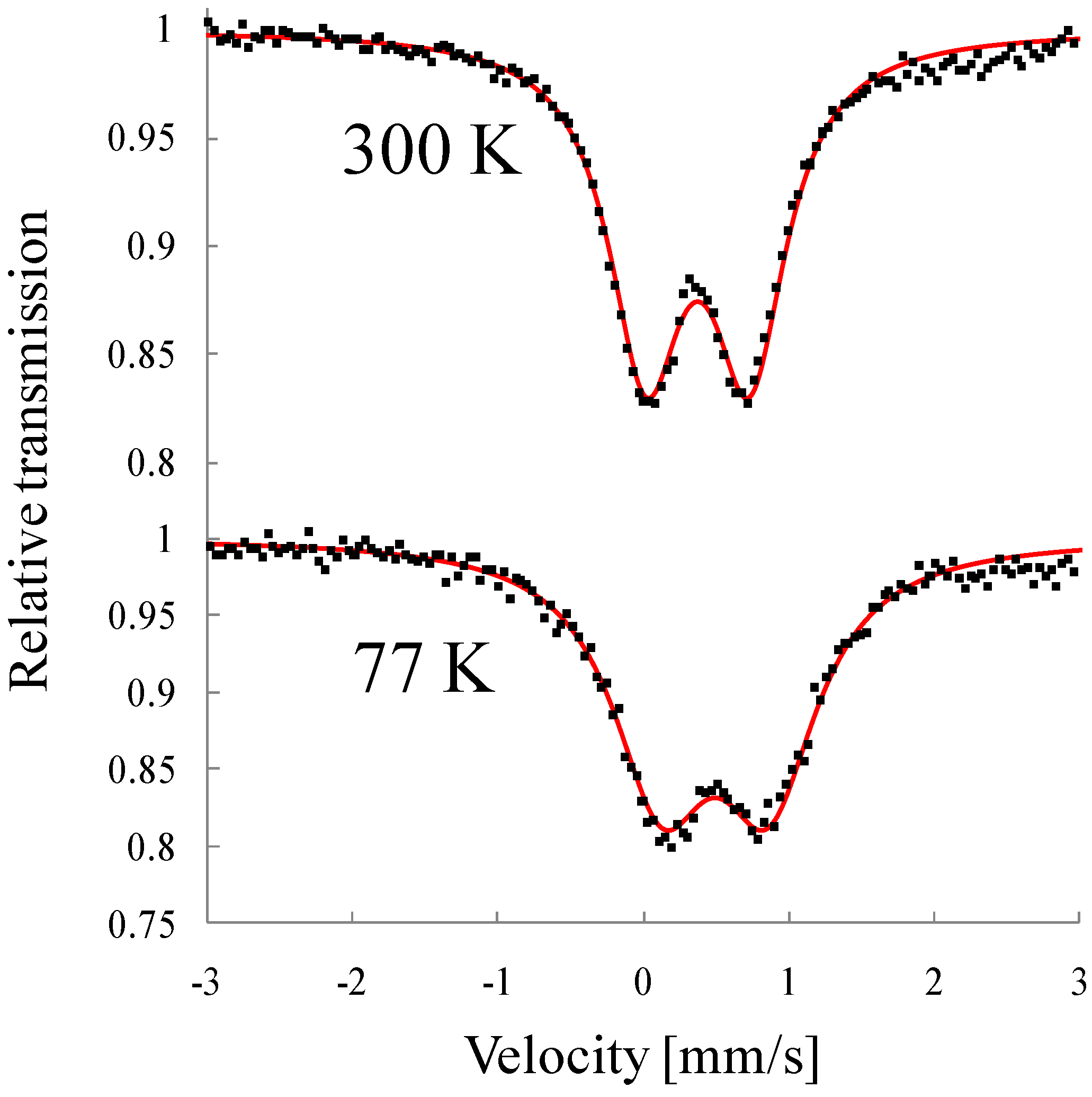
Figure 20. The line profiles of both complexes are quite similar to each other and two quadrupole doublets are assigned to Fe
II(
S = 2) and Fe
III(
S = 1/2). The values of the isomer shifts and quadrupole splittings of SP-Me are very close to those of (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3] with the spin states of Fe
II(
S = 2) and Fe
III(
S = 1/2) [
39]. Moreover, the infrared spectra indicate that all of the dithiooxalate groups in these complexes act as bridging ligands [
68].
Figure 20.
57Fe Mössbauer spectra of (
a) (SP-Me)[Fe
IIFe
III(dto)
3] and (
b) (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at room temperature [
42].
Figure 20.
57Fe Mössbauer spectra of (
a) (SP-Me)[Fe
IIFe
III(dto)
3] and (
b) (
n-C
3H
7)
4N[Fe
IIFe
III(dto)
3] at room temperature [
42].
According to the infrared and Mössbauer spectra, the 2D honeycomb network structure of [FeIIFeIII(dto)3] layer is formed in SP-Me complex, where the FeII(S = 2) and FeIII(S = 1/2) sites are coordinated by six O atoms and six S atoms, respectively.
2.6.2. Photoisomerization in solid state
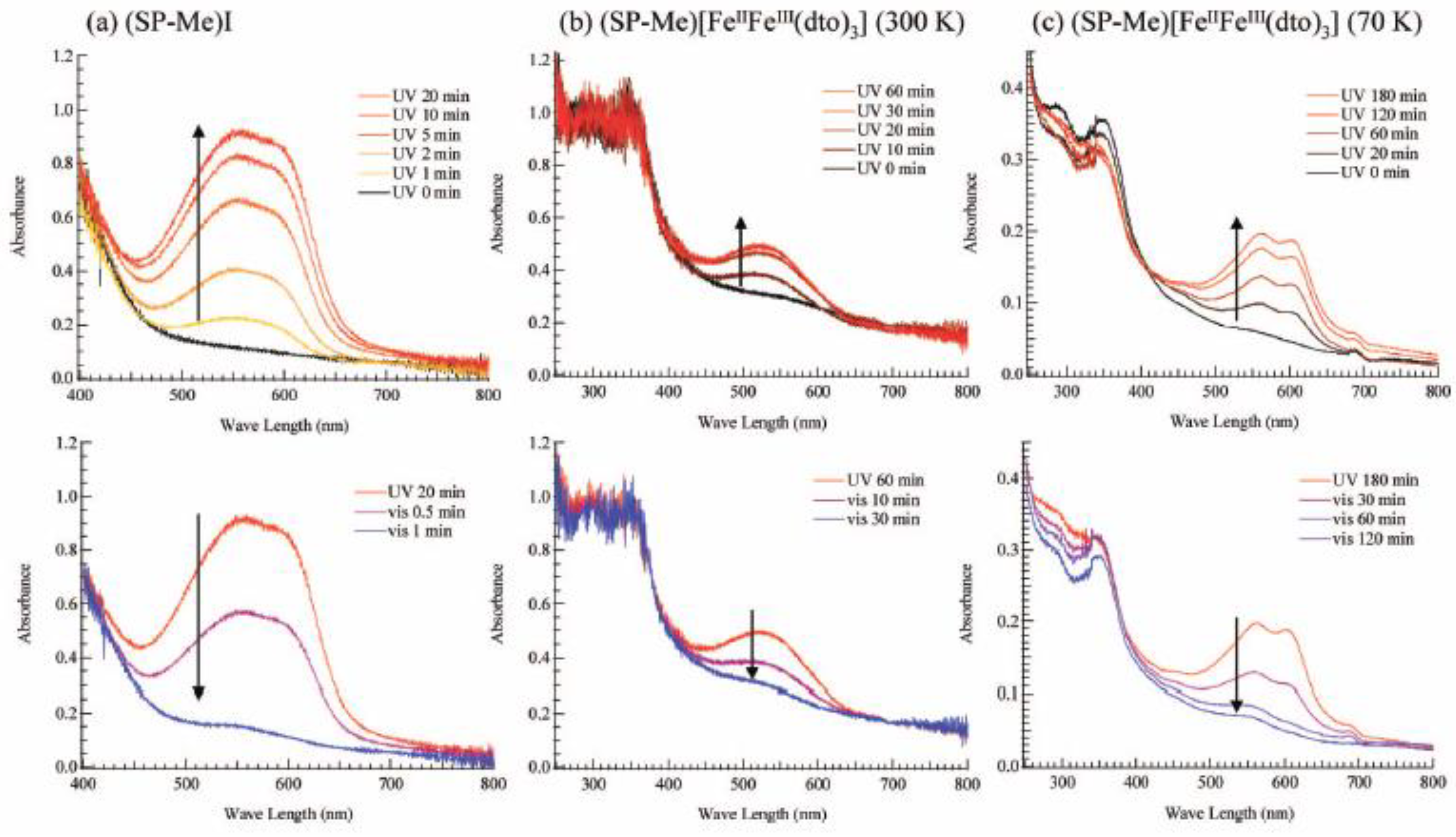
Figure 21(a) shows the UV-vis absorption spectra for (SP-Me)I in a KBr pellet. Upon UV irradiation at 350 nm, a broad absorption band between 500 and 650 nm, corresponding to the π-π* transition of the OF, appears. In contrast, the visible light irradiation of 570 nm returned the saturated spectrum to the original one. This photochromism is based on the UV-vis light-induced equilibrium between the yellow-colored closed form (CF) and the red-colored open form (OF).
Figure 21(b) and (c) show the changes of the absorption spectra for SP-Me complex in KBr pellet upon UV irradiation at 350 nm at 300 and 70 K, respectively. In the case of 300 K, the intensity of the top of the peak around 570 nm is continuously enhanced with the increase of UV irradiation time while the initial black pellet turns deep purple. After the UV irradiation for 30 min, the intensity of the absorption spectrum corresponding to the π-π* transition of the OF is almost saturated. The UV light-induced OF of SP-Me complex is stable even at room temperature in the dark condition and the purple color slowly fades to return to black in several days. In contrast, visible light irradiation considerably accelerates the color decay. This result implies that the photoisomerization of cationic SP-Me molecule from CF to OF by UV irradiation and from OF to CF by visible-light irradiation reversibly takes place in the solid state of SP-Me complex at room temperature. At 70 K, the photoisomerization of cationic SP-Me molecule is also induced by UV irradiation, and the intensity of the absorption spectrum around 570 nm is almost saturated in 180 min. In this case, the absorption band also almost disappears upon visible-light irradiation for 120 min.
Figure 21.
UV-vis spectral change of (
a) (SP-Me)I and (SP-Me)[Fe
IIFe
III(dto)
3] in KBr pellet at (
b) 300 and (
c) 70 K. UV irradiation (350 nm, 40 mW/cm
2) on the pellet was carried out first. After the spectra were saturated, visible-light irradiation (600 mW/cm
2) was carried out on the pellet [
42].
Figure 21.
UV-vis spectral change of (
a) (SP-Me)I and (SP-Me)[Fe
IIFe
III(dto)
3] in KBr pellet at (
b) 300 and (
c) 70 K. UV irradiation (350 nm, 40 mW/cm
2) on the pellet was carried out first. After the spectra were saturated, visible-light irradiation (600 mW/cm
2) was carried out on the pellet [
42].
2.6.3. Photocontrollable CTPT and ferromagnetic transition
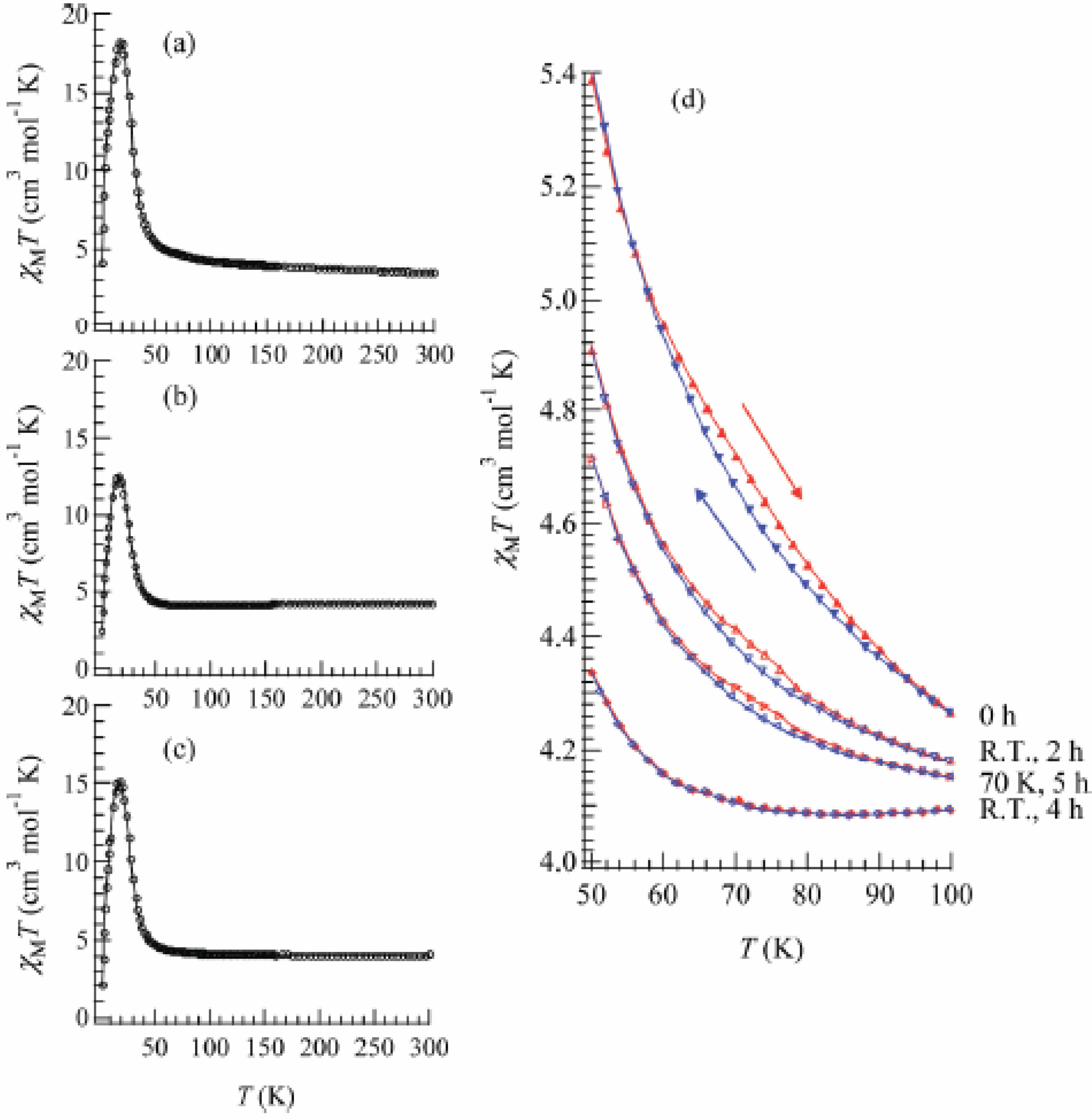
Figure 22 shows the temperature dependence of the product of the molar magnetic susceptibility and temperature, χ
MT of the SP-Me complex. The effective moment at room temperature is 5.28 μ
B which is close to that of the spin only value of the high temperature phase (HTP) with Fe
II (
S = 2) and Fe
III (
S = 1/2) (5.20 μ
B,
g = 2). The Curie constant and Weiss constant calculated from the value above 100 K are 3.25 cm
3·mol
-1·K and +26.2 K, respectively. It is remarkable that the χ
MT curve exhibits a thermal hysteresis loop between 50 and 100 K showing a small bump around 75 K as shown in
Figure 22(d), which indicates that the CTPT occurs as well as in the case of (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3] (n = 3 and 4) in the similar temperature range. As the temperature is lowered below 50 K, χ
MT rapidly increases up to a maximum value around 18 K and the magnetization is saturated below that temperature, which suggests that the SP-Me complex exhibits a long-range ferromagnetic ordering as well as (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3].
Figure 22.
Temperature dependence of χ
T of (SP-Me)[Fe
IIFe
III(dto)
3] before (
a) and after UV irradiation (350 nm, 40 mW/cm
2) at (
b) 300 K and (
c) 70 K. Applied magnetic field was 5000 Oe. (
d) UV irradiation effect on the χ
T versus T in the temperature range around the charge transfer phase transition [
42].
Figure 22.
Temperature dependence of χ
T of (SP-Me)[Fe
IIFe
III(dto)
3] before (
a) and after UV irradiation (350 nm, 40 mW/cm
2) at (
b) 300 K and (
c) 70 K. Applied magnetic field was 5000 Oe. (
d) UV irradiation effect on the χ
T versus T in the temperature range around the charge transfer phase transition [
42].
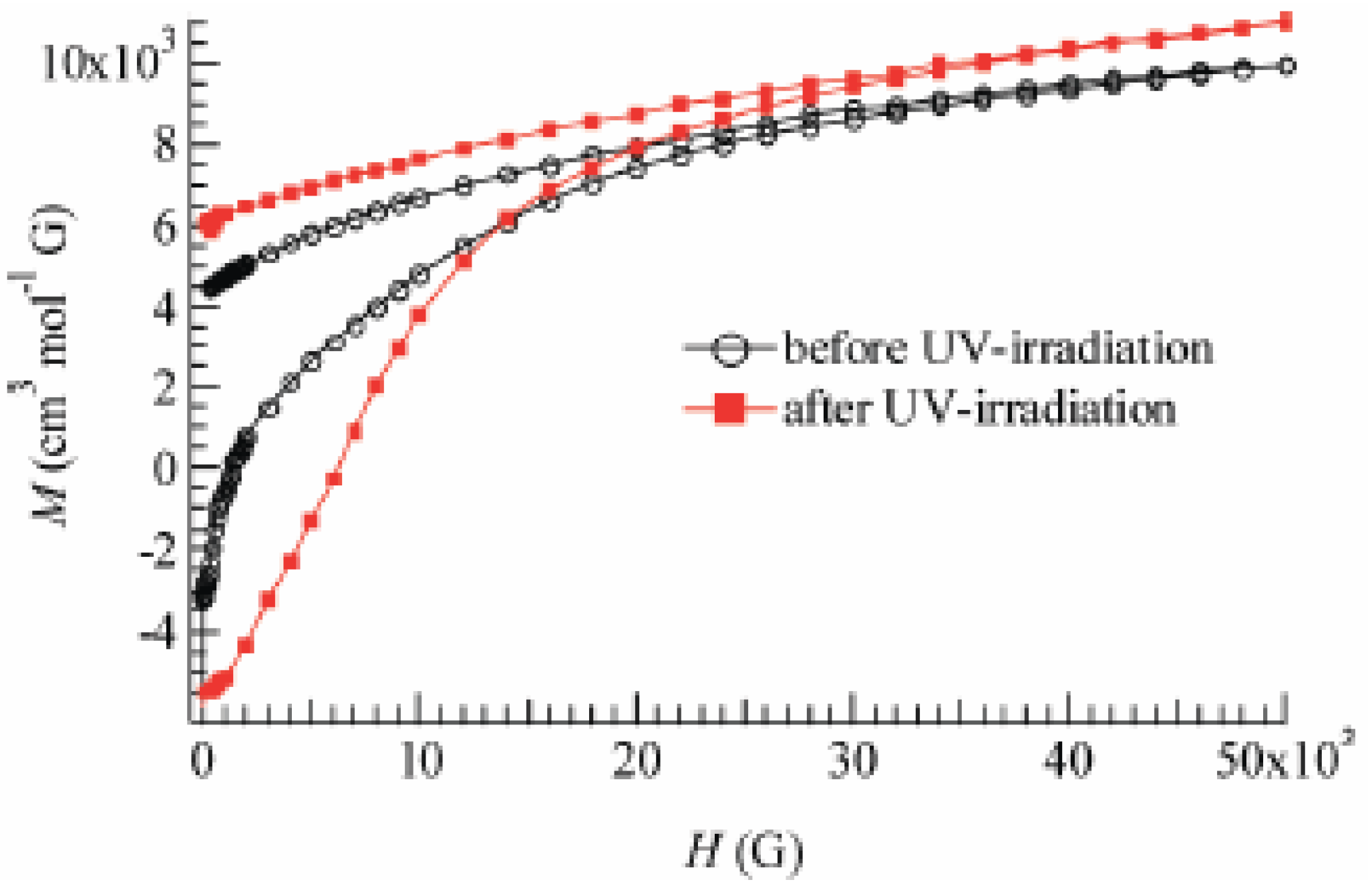
To confirm and characterize the ferromagnetically ordered phase, the FCM, RM, ZFCM and the field dependence of the magnetization at 2 K were measured. These results are displayed in
Figure 23 and
Figure 24. The FCM curve decreases stepwise at 7 K and disappears at about 25 K.
Figure 23.
Temperature dependence of FCM, RM, and ZFCM of (SP-Me)[Fe
IIFe
III(dto)
3] before
(a) and after UV irradiation (350 nm, 40 mW/cm
2) at 300 K (
b and
c) and 70 K (
d). Applied magnetic field is 30 Oe [
42].
Figure 23.
Temperature dependence of FCM, RM, and ZFCM of (SP-Me)[Fe
IIFe
III(dto)
3] before
(a) and after UV irradiation (350 nm, 40 mW/cm
2) at 300 K (
b and
c) and 70 K (
d). Applied magnetic field is 30 Oe [
42].
Figure 24.
Field dependence of the magnetization of (SP-Me)[Fe
IIFe
III(dto)
3] at 2 K before and after UV irradiation (350 nm, 40 mW/cm
2) at room temperature [
42].
Figure 24.
Field dependence of the magnetization of (SP-Me)[Fe
IIFe
III(dto)
3] at 2 K before and after UV irradiation (350 nm, 40 mW/cm
2) at room temperature [
42].
As shown in
Figure 23(a), the RM curve also decreases stepwise at 7 K and then disappears at about 22 K. The ZFCM curve, on the other hand, has two maxima at 5 and 18 K. This peculiar behavior of magnetization curves is quite similar to that of (
n-C
4H
9)
4N[Fe
IIFe
III(dto)
3] in which the LTP and HTP coexist even in the temperature region below the CTPT. In analogy with (
n-C
4H
9)
4N[Fe
IIFe
III(dto)
3], the LTP and HTP of the SP-Me complex individually undergo the ferromagnetic phase transitions with
TC(LTP) = 5 K and
TC(HTP) = 22 K, respectively. The field dependence of the magnetization at 2 K exhibits the hysteresis loop characteristic of ferromagnetic materials with a coercive field of 1400 Oe. The magnetization at
H = 50000 Oe yields about 1.90 μ
B.
Moreover, the ferromagnetism of the SP-Me complex shows a noteworthy response upon UV irradiation.
Figures 23(b) and (c) show the FCM, RM, and ZFCM curves of the SP-Me complex after UV irradiation of 350 nm at room temperature. The magnetization value below 7 K starts decreasing upon the UV irradiation. The steps in the FCM and RM at 7 K are lowered, and the peak around 5 K in ZFCM disappears, indicating the disappearance of the LTP. On the other hand, the magnetization values between 7 and 30 K are slightly increased after the UV irradiation. Moreover, the thermal hysteresis loop in χ
T versus T plot gradually vanishes with the UV irradiation, which is shown in
Figure 22(d). This photo-induced effect can be explained by the suppression of the CTPT. The photoisomerization of cationic SP-Me molecule in the SP-Me complex leads to the expansion of its own volume, which gives a significant stress to the framework of [Fe
IIFe
III(dto)
3] layers and expands the unit cell volume.
Taking into account that the CTPT in the (n-CnH2n+1)4N[FeIIFeIII(dto)3] series tends to be inhibited by the expansion of their (n-CnH2n+1)4N+ cation size, it can be concluded that the HTP in the SP-Me complex becomes more stable than the LTP between 2 and 300 K through the medium of the photoisomerization of cationic SP-Me molecule. In order to elucidate the mechanism of the photo-induced effect on the magnetic properties of SP-Me complex, we carried out the control experiment for the magnetism of (n-C4H9)4N[FeIIFeIII(dto)3] whose intercalated cation has no photochromic property. After 4 h of UV irradiation (350 nm) at room temperature, the magnetization of (n-C4H9)4N[FeIIFeIII(dto)3] shows no change. This result gives strong evidence that the disappearance of the LTP in the SP-Me complex after UV irradiation is caused by the photoisomerization of cationic SP-Me molecule.
Figure 24 also shows the field dependence of the magnetization at 2 K after 4 h of UV irradiation at room temperature. The coercive force at 2 K is enhanced from 1400 to 6000 Oe after the UV irradiation. In connection with this, the coercive forces of (
n-C
nH
2n+1)
4N[Fe
IIFe
III(dto)
3] at 2 K are 310, 3160, 6600 and 6800 Oe for n = 3, 4, 5, and 6, respectively [
40], suggesting the HTP has higher coercive force than the LTP. This result supports the photo-induced HTP giving rise to long-range magnetic ordering. Before UV irradiation, the CTPT occurs around 75 K, while the LTP and HTP coexist below that temperature. There seems to be two phases in the SP-Me complex from the analysis of the temperature dependence of the magnetic susceptibility. Here we name these phases A and B respectively. The A phase undergoes the CTPT, and the HTP(A) perfectly converts into the LTP(A) with decreasing temperature. On the other hand, the B phase does not undergo the CTPT, and the HTP(B) is more stable than the LTP(B) in the whole temperature range. The reaction scheme of the SP-Me complex is illustrated in
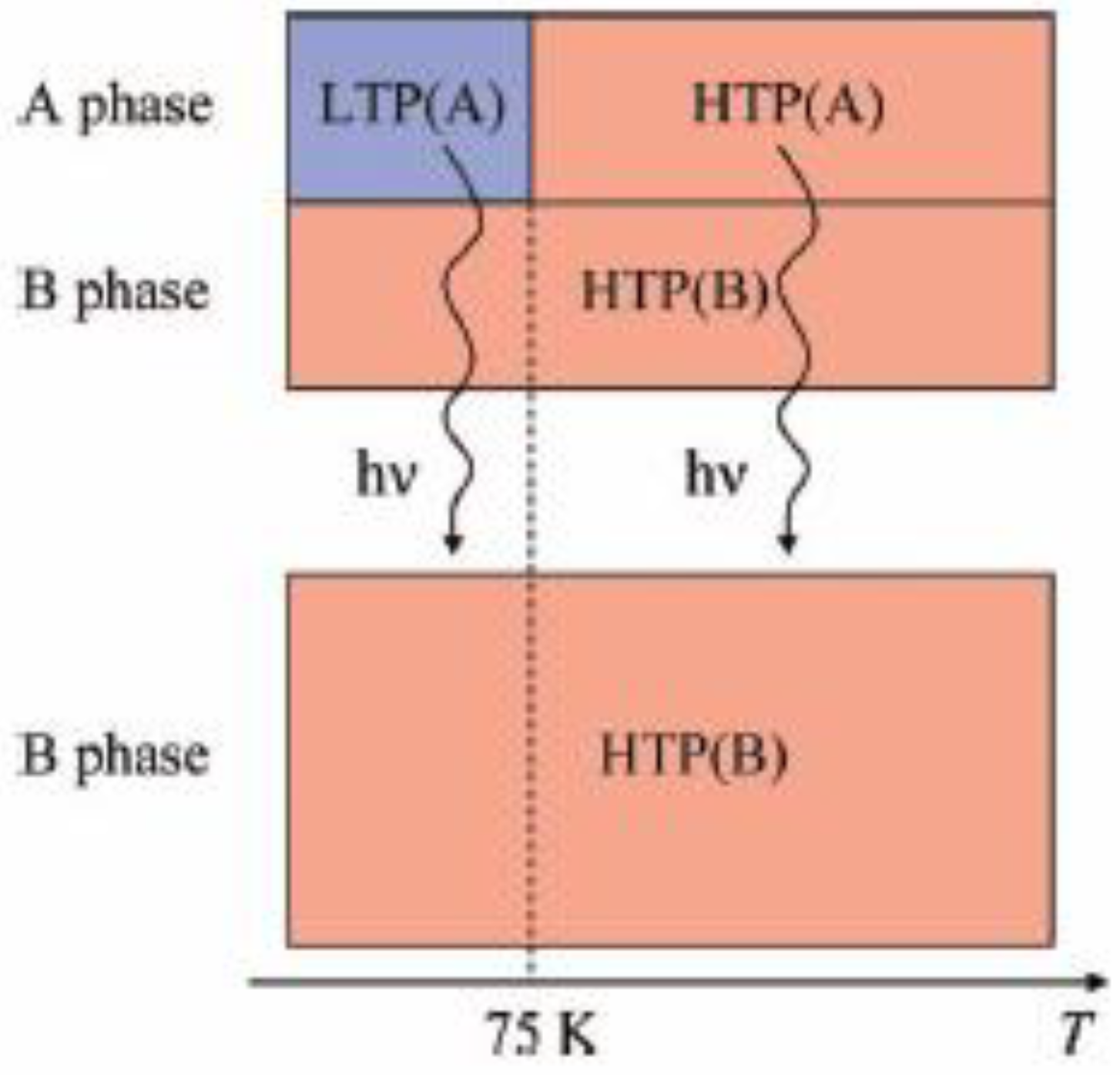
Figure 25. Suppose that the UV irradiation induces the transformation from the A phase to the B phase, the LTP(A) can be forced to convert into the HTP(B) by UV irradiation below 90 K,
i.e., the photoisomerization-induced CTPT schematically shown in
Figure 26 is realized. Here, 90 K is the upper limit of the thermally induced CTPT in the SP-Me complex. In order to prove the concerted phenomenon coupled with the CTPT in [Fe
IIFe
III(dto)
3] and the photo-isomerization of the intercalated SP-Me molecule in the SP-Me complex, we performed a low-temperature irradiation experiment which corresponds to the arrow of the left side in
Figure 25. The FCM, RM, and ZFCM curves of SP-Me complex after the UV irradiation at 70 K are shown in
Figure 23(d), and the χ
T versus
T plot of the SP-Me complex after the UV irradiation is shown in
Figure 22(c). In fact, the photo-induced change in the magnetic property displays the same tendency as in the case of UV irradiation at 300 K. It should be noted that the destabilization of LTP and the stabilization of HTP under UV irradiation below 90 K induces the CTPT in the [Fe
IIFe
III(dto)
3] layers. This result proves that the photoisomerization-induced CTPT is realized in this organic-inorganic hybrid system.
Figure 25.
Reaction scheme of (SP-Me)[FeIIFeIII(dto)3] before and after UV irradiation.
Figure 25.
Reaction scheme of (SP-Me)[FeIIFeIII(dto)3] before and after UV irradiation.
Figure 26.
Schematic representation of the concerted phenomenon coupled with the charge transfer phase transition in [FeIIFeIII(dto)3] and the photoisomerization of spiropyran in (SP-Me)[FeIIFeIII(dto)3]. White colored and black colored circles are the FeII and FeIII sites, respectively.
Figure 26.
Schematic representation of the concerted phenomenon coupled with the charge transfer phase transition in [FeIIFeIII(dto)3] and the photoisomerization of spiropyran in (SP-Me)[FeIIFeIII(dto)3]. White colored and black colored circles are the FeII and FeIII sites, respectively.
This new type of photomagnetism coupled with spin, charge and photon is triggered by a chemical pressure effect generated from the photoisomerization of spiropyran from the closed form to the open one in the complex. This situation seems to have significant similarity with the first events in the perception of light in rhodopsin in which photoisomerization of 11-cis-retinal into all-trans-retinal induces a conformational change in opsin and activates the associated G protein and triggers a second messenger cascade.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





































