1. Introduction
Computer simulations on biological macromolecules have been a hot area for numerous theoretical studies describing biological processes at the molecular level. One of the popular treatments for this purpose is the classical molecular dynamics simulation method in which empirical interatomic potential functions are used. However, it is not so accurate because of the treatment of constant atom charges. On the other hand, in
ab initio molecular dynamics (AIMD), the electronic structure, energy and forces of the system are directly computed based on quantum mechanics theory. However, the number of atoms one can handle in the quantum mechanical treatment is much smaller than the number of atoms in a biological system because of the limitation of computer resources. The elongation (ELG) method has proved to be an efficient method for calculating the electronic structure of large aperiodic polymers with high accuracy at the
ab initio level [
1,
2,
3,
4,
5,
6]. In addition, the analytical energy gradients can be calculated by the ELG method and have been successfully used for the geometry optimization of a series of polymers [
7]. Based on these developments, we here propose a new efficient AIMD method named Elongation-MD (ELG-MD) by combining the elongation method with the dynamics algorithm. The ELG-MD method makes it possible to efficiently analyze the mechanisms of chemical reactions by considering dynamics even for huge random polymers such as biomaterials. For the first application of the ELG-MD method to confirm its effectiveness, we selected polyglycine as a target system; polyglycine is representative of the simplest peptide and widely investigated both experimentally and theoretically.
Polyglycine (Gly)
n, containing n residues, forms the backbones of amino acids, peptides and proteins without side chains and their functional groups [
8]. Therefore, the investigations on (Gly)
n are important to the understanding of a wide range of biological systems. In particular, the three-dimensional structure is one key factor for the biological activities of a peptide, and hydrogen bonds play important roles in stabilizing the secondary structure of peptides [
9]. In general, hydrogen bonds (H-bonds) can be generated both intramolecularly and intermolecularly in the system. In the former case, the H-bonds are formed between many different functional groups in the peptide, but in the latter case the H-bonds are formed between polar groups of the peptide and the surrounding solvent molecules such as water molecules. For a β strand, the hydrogen bonds are formed by the N–H groups in the backbone of one β strand with the C=O groups in the backbone of the adjacent β strands. For an α helix, each hydrogen bond is formed by the N–H group of an amino acid with the C=O group of the amino acid four residues earlier. On the conformation of polyglycine, Ohnishi
et al. demonstrated that polyglycine in solution exhibits a strong preference for an extended conformation when polymerized in short segments as the intermediate in the extension between a β strand and an α helix [
10]. Which kind of hydrogen bond will stabilize this extended conformation of polyglycine in aqueous solution? The hydration effects via direct hydrogen bonds between water molecules and the peptide backbone can be expected. Here we applied the ELG-MD method to investigate the intermolecular H-bonds effects on conformation stability when (Gly)
n is polymerized with water molecules. This application deals with the conformation change of (Gly)
n under the explicit consideration of water molecules, and the AIMD simulation in which energies and forces are generated by the ELG method at each time step.
This paper is organized into four sections. In
Section 2, we briefly introduce the ELG formalism including energy and gradient calculations, after that we present a description of the ELG-MD method. In
Section 3, we introduce the structure of model molecules,
i.e., polyglycine with water molecules. In the same section we analyze the equilibrium structures of polyglycine and their energetic stability. Final conclusions are collected in
Section 4.
3. Results and discussion
A (Gly)
14 peptide in β-strand conformation with 14 water molecules was selected as a model system to test and demonstrate the ELG-MD method (
Figure 3). For the first testing calculation for ELG-MD, we did not consider more water molecules around polyglycine. One reason was to save computing time, another reason is that for the central fragment of the glycine molecule, one side including C=O and N–H groups is hydrophilic and these two groups can form a hydrogen bond ring C
7(C=O···H–O···H–N) (shown in
Figure 4b) with one water molecule, it is not easy to add more water molecules forming hydrogen bonds with these two groups in this area because of space limitation. Another side including the CH
2 group is hydrophobic. Here, the notation C
n (X···H–O···Y) represents the hydrogen bond ring structure consisting of n atoms enclosed by the H–O group of the adjacent water molecule. The (Gly)
14 is divided into seven units ((Gly)
2 for each unit), and assigned A, B, and M
1~M
3 regions as shown in
Figure 3. The simulation in the present article has two stages.
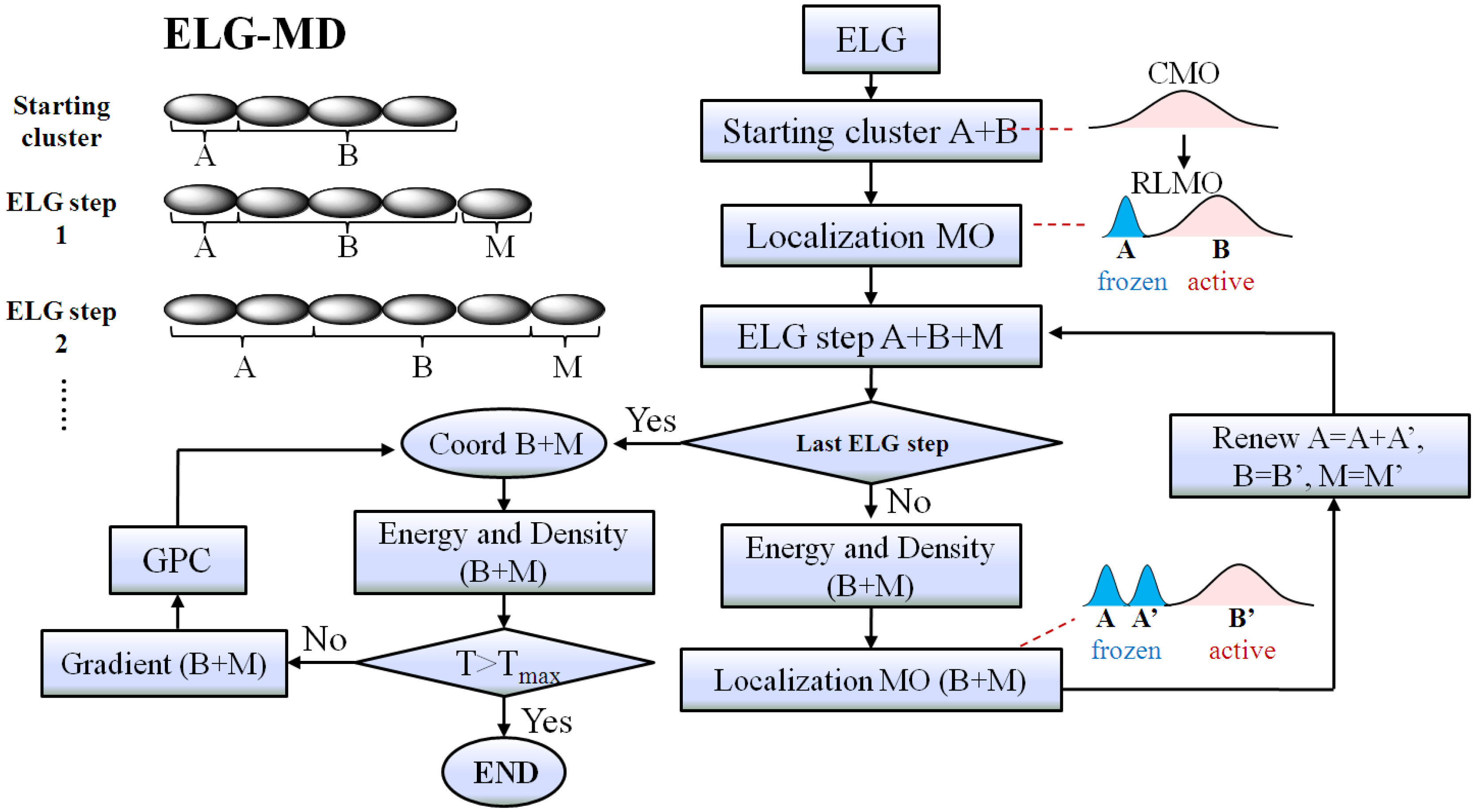
Figure 2.
The flowchart of the elongation molecular dynamics (ELG-MD) method. The “Energy and Density (B + M)” means that the energy and density of the B + M region are obtained with the contribution from the A region.
Figure 2.
The flowchart of the elongation molecular dynamics (ELG-MD) method. The “Energy and Density (B + M)” means that the energy and density of the B + M region are obtained with the contribution from the A region.
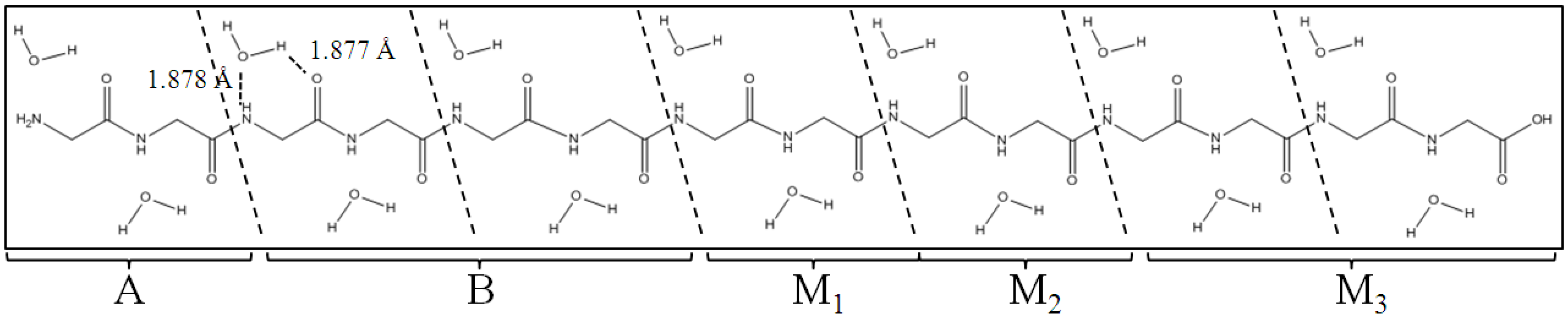
Figure 3.
Polyglycine (Gly)
14 in β-strand conformation with 14 water molecules. (Gly)
14 are divided into A, B, M
1, M
2 and M
3 regions for the ELG-MD procedures. The notations A and B do not correspond to those in
Figure 2.
Figure 3.
Polyglycine (Gly)
14 in β-strand conformation with 14 water molecules. (Gly)
14 are divided into A, B, M
1, M
2 and M
3 regions for the ELG-MD procedures. The notations A and B do not correspond to those in
Figure 2.
First, we need a stable conformation of A, B, M
1, and M
2 regions to create a more real surrounding for the new attacking monomer (M
3 region) before investigating the structure of (Gly)
14 including the A, B, and M
1~M
3 regions. Several research groups have devised experiments for measuring β-sheet stability with various peptides, and they found polyglycine has a low β-sheet propensity [
17,
18,
19,
20]. Therefore, to find a more reasonable conformation than β-sheet conformation, the conventional AIMD simulation was performed for the A, B, M
1 and M
2 regions (other than the M
3 region). In the AIMD simulation, the energies and forces were obtained at the HF level of theory with the STO-3G basis set. The time step is set as 0.5 fs and we perform in total 5 ps simulation (10,000 steps). The temperature of the system is associated with the classical kinetic energy and the constant temperature (298 K) is achieved by the algorithm proposed by Berendsen
et al. [
21].
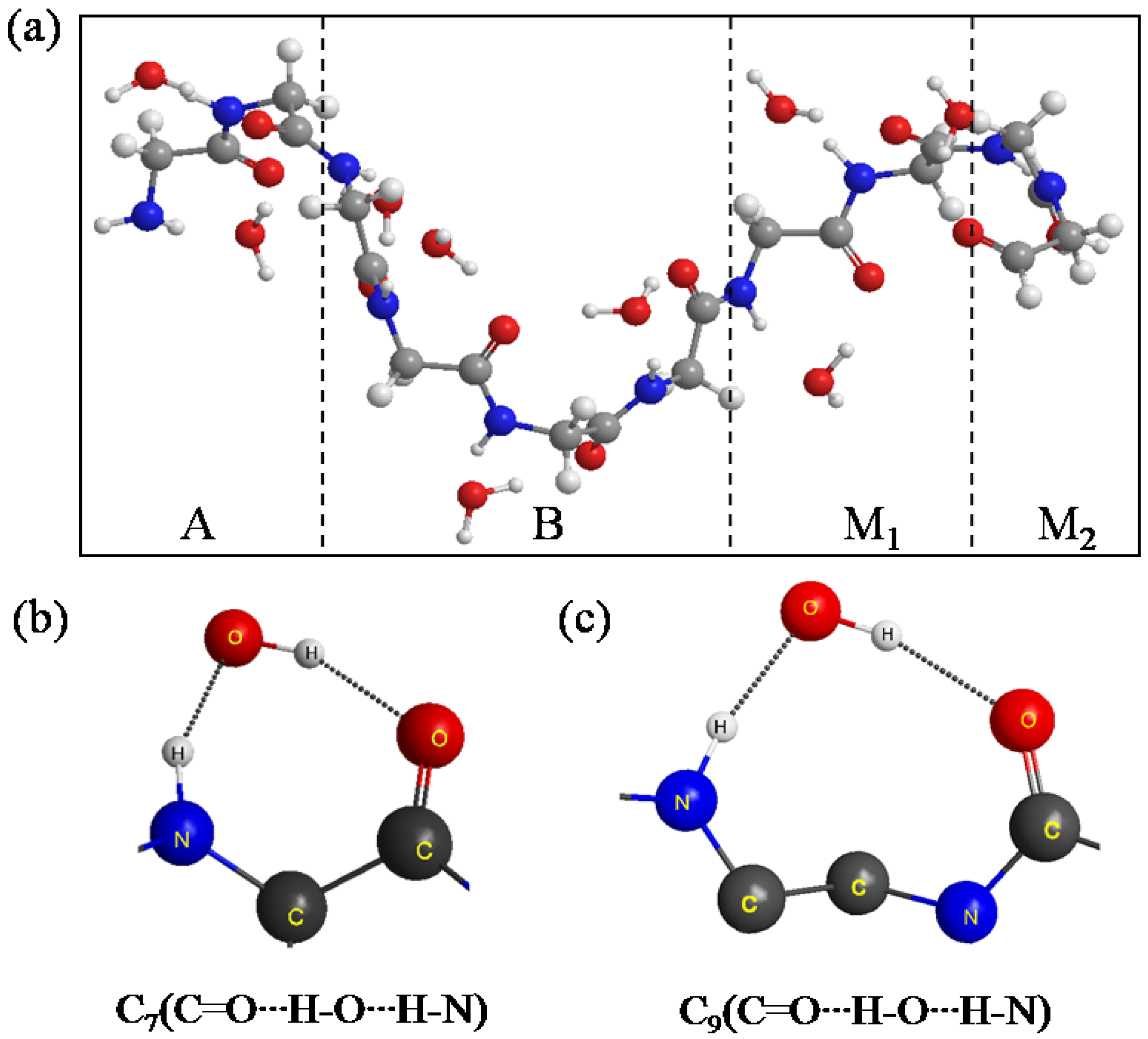
Figure 4a shows the equilibrium structure obtained by the simulation. It was found that the conformation of A, B, M
1 and M
2 significantly changed from a β-sheet type to a quasi-α-helix type in which the structure is more outstretched than α-helix. In general, α-helix forms a right-handed helix and every N–H group in the backbone donates a hydrogen bond to the C=O group of the amino acid belonging to four residues earlier (I + 4 → i hydrogen bonding) [
22]. Our results in
Figure 4a show that the right-handed helix is induced by two kind of hydrogen bond rings, that is, C
7(C=O···H–O···H–N) in
Figure 4b and C
9(C=O···H–O···H–N) in
Figure 4c. C
9(C=O···H–O···H–N) shows that the H–O group of the water donates two hydrogen bonds to the C=O and N–H groups of the backbone for instance.
Figure 4.
(a) The structure of the A, B, M1 and M2 regions of (Gly)14 in a quasi-α-helix conformation as the initial structure for ELG-MD simulation. Panels (b) and (c) show two types of H-bond ring. The dotted lines in the panels denote hydrogen bonds.
Figure 4.
(a) The structure of the A, B, M1 and M2 regions of (Gly)14 in a quasi-α-helix conformation as the initial structure for ELG-MD simulation. Panels (b) and (c) show two types of H-bond ring. The dotted lines in the panels denote hydrogen bonds.
Second, for the ELG-MD calculation, there are four calculation steps including starting cluster (A and B regions), the first elongation step (A, B and M
1 regions), the second elongation step (A, B, M
1 and M
2 regions) the third elongation step (A, B, M
1, M
2 and M
3 regions). Energy calculations are carried out for the first three steps, and MD only performed for the last step. In the last step, we presume that a new monomer M
3 (β form) furthermore attacks the propagating site of the M
2 region of the structure obtained above. Therefore, the initial structure for the ELG-MD method consists of the A + B + M
1 + M
2 equilibrium structure (quasi-α form) and an additional M
3 region (β form). The M
1~M
3 region of the initial structure is shown in
Figure 5. Because we are interested in the structure variation of M
3 and the associated regions such as the M
1, M
2 regions, the MD treatment in the ELG-MD procedure is applied only for the interacting space in the last elongation step, that is, the M
1~M
3 region while the A and B regions are frozen. For the dynamics part of the ELG-MD calculation, we used the same setting as conventional AIMD simulation.
To know the efficiency of the ELG-MD method, 2.5 ps (5000 steps) simulation was performed by both ELG-MD and conventional AIMD for the initial structure in
Figure 5. The total wall clock time is 206,362.3 s (ELG-MD) and 298,391.3 s (conventional AIMD), respectively. Because the ELG-MD is a local MD method, its efficiency is obviously higher (about 31%) than the conventional AIMD. Furthermore, the potential energies (HF energies) of the entire system can be compared between the two treatments (see
Figure 6).
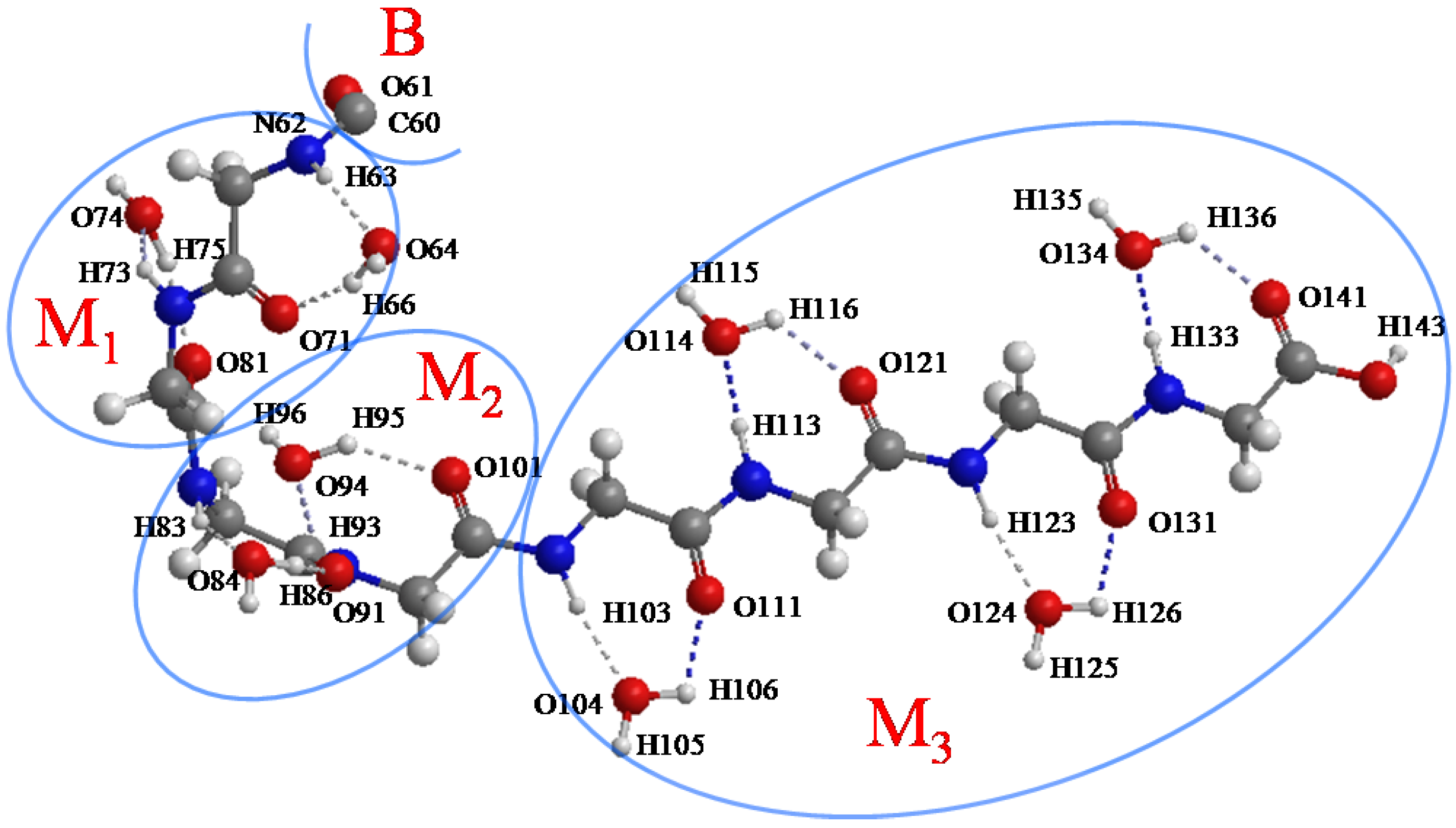
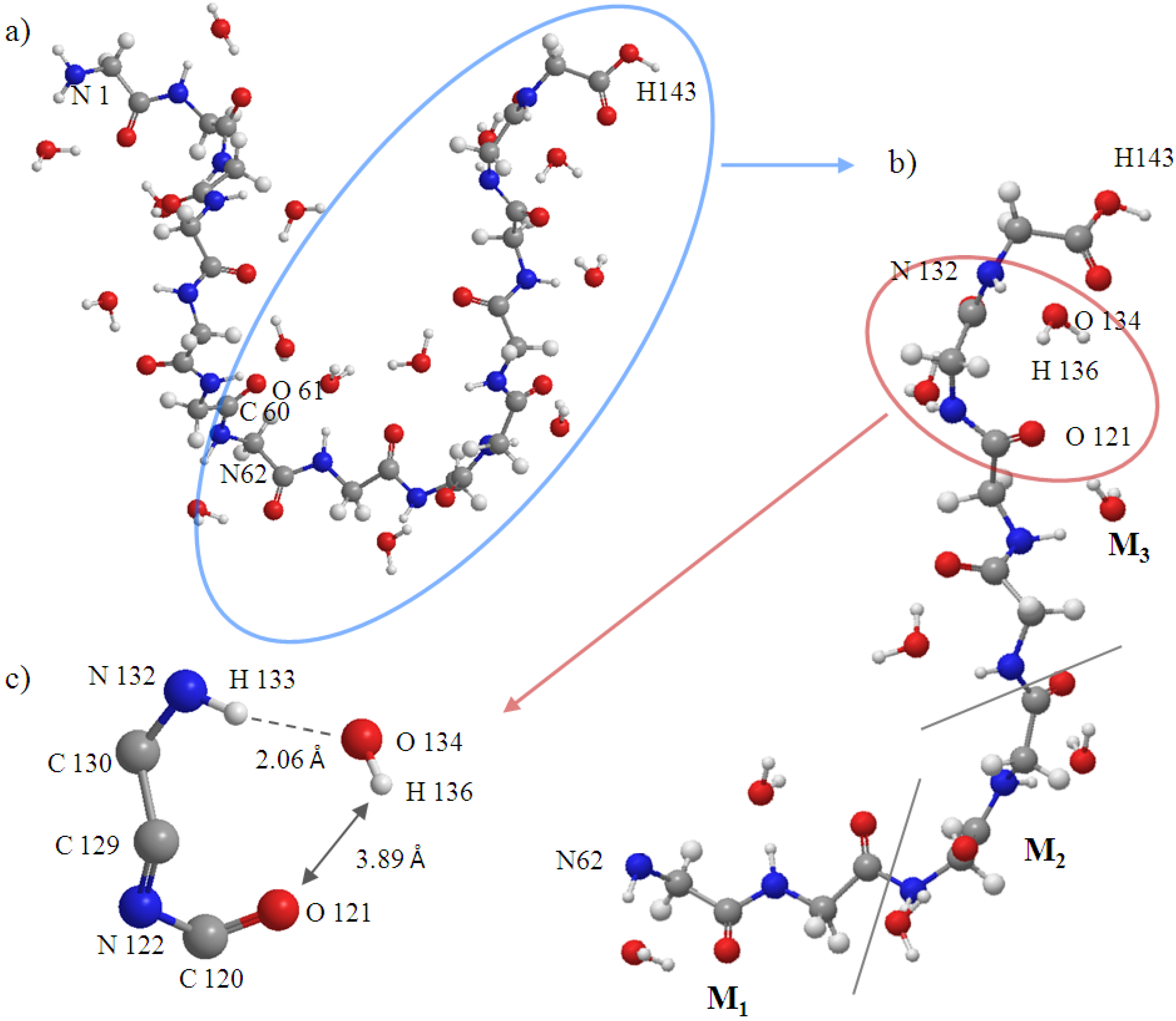
Figure 5.
The initial structure of the M1, M2 and M3 regions (atoms from 62 to 143). C60 and O61 belong to the B region.
Figure 5.
The initial structure of the M1, M2 and M3 regions (atoms from 62 to 143). C60 and O61 belong to the B region.
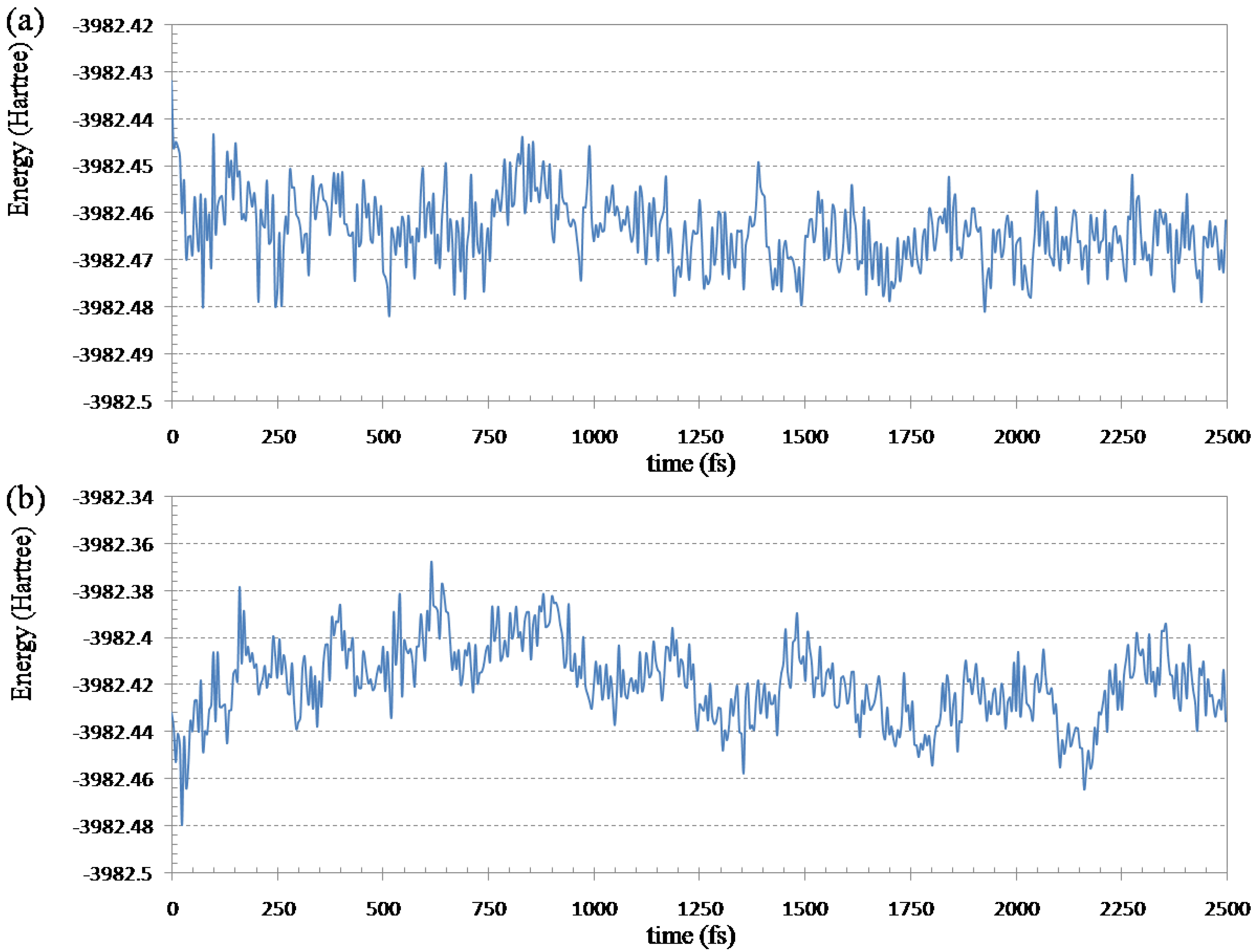
Figure 6.
Fluctuations of the Hartree-Fock energy of (Gly)14 with 14 water molecules in the simulation at 298.15 K: (a) ELG-MD simulation; (b) conventional ab initio MD simulation.
Figure 6.
Fluctuations of the Hartree-Fock energy of (Gly)14 with 14 water molecules in the simulation at 298.15 K: (a) ELG-MD simulation; (b) conventional ab initio MD simulation.
From
Figure 6a we can see the HF energy decreases quickly for the ELG-MD method which relaxes only a local region of the polymer. In the ELG-MD calculation, the structure of the A and B regions is automatically frozen when the M
3 region attaches, which is different from the conventional AIMD which treats the whole system at the same time. Because the freedom of the ELG-MD calculation is much less than conventional AIMD, it is much easier for the ELG-MD method to get the energy convergence.
Figure 6b shows the fluctuations of the Hartree-Fock energy calculated by conventional AIMD. The energy increases during 900 fs, and then the energy begins to decrease slowly. That is to say, it is hard to get the energy equilibrium in a short period of time in the conventional AIMD. It also should be stressed that the energy of ELG-MD is lower than that of conventional AIMD at 2500 fs. From 2000 to 2500 fs, the average total energy is −3982.4664 Hartree for ELG-MD while −3982.4245 Hartree for AIMD. When the new monomer attaches to the propagating site of the M
2 region, it will mainly affect the structure of the M
1 and M
2 regions because the A and B regions are far away from the M
3 region (the distance between them is about 10 Å). Therefore, we only focus on the conformation variation of the attacking monomer (M
3) and its vicinity (M
1 and M
2) under the assumption that the A and B regions have no interactions with the M
3 region.
To check the accuracy of the ELG-MD, we performed 0.5 ps (1000 steps) conventional AIMD calculation for the polymer with fixing of the atoms in the A and B regions in the same manner as with the ELG-MD calculation. We compared the structures of them in each simulation step.
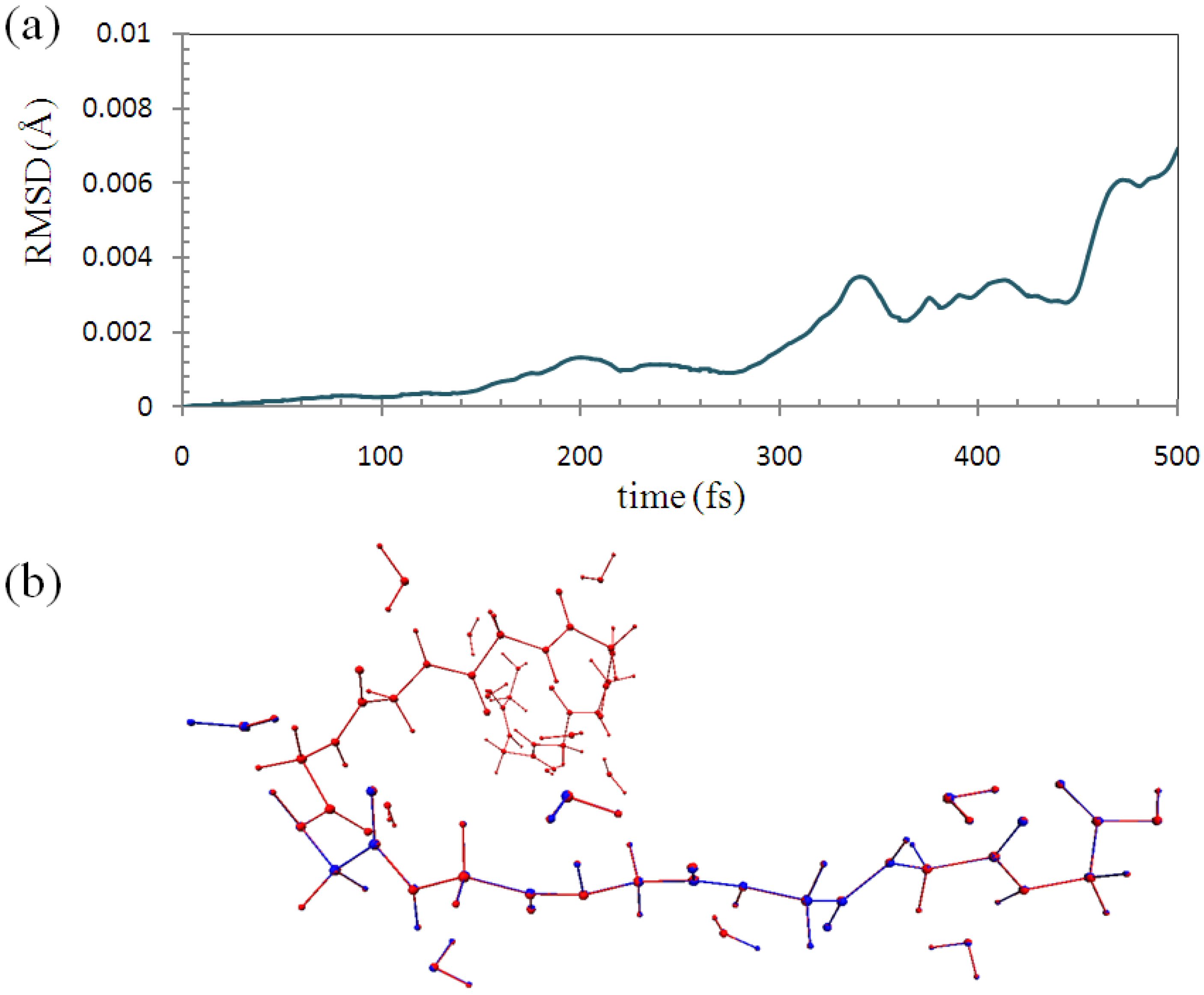
Figure 7a shows the root-mean-square deviation (RMSD) of the backbone part (M
1~M
3) of the structure between the ELG-MD and conventional AIMD with fixing the atoms. The maximum value of RMSD with 0.00695 Å is considerably small. This shows that the two structures agree well in each simulation step.
Figure 7.
(a) Root-mean-square deviation (RMSD) of the molecular structure between ELG-MD and conventional ab initio MD with fixing of the same atoms; (b) Superimposed structures at 500 fs. (Red: conventional AIMD. Blue: ELG-MD).
Figure 7.
(a) Root-mean-square deviation (RMSD) of the molecular structure between ELG-MD and conventional ab initio MD with fixing of the same atoms; (b) Superimposed structures at 500 fs. (Red: conventional AIMD. Blue: ELG-MD).
As shown in
Figure 7b, the two structures at 500 fs (1000 steps) calculated by the two methods are almost the same as each other. Hence, it can be concluded that the trajectories of the two simulations have essentially the same quality, and the structure obtained by the ELG-MD simulation is credible.
Now, we performed 5 ps (10,000 steps) ELG-MD simulation for the (Gly)
14 peptide starting from the initial structure in
Figure 5 to investigate the conformation variation from the viewpoint of the H-bond effects between peptide and water molecules. It should be noted that the frozen region (A and B) is not shown in
Figure 5 because the coordinates of the frozen regions are fixed. As mentioned above, in the initial structure the M
3 region has β-strand confirmation and the other region A + B + M
1 + M
2 has quasi-α-helix conformation. All of the H-bonds in the initial structure are of the C
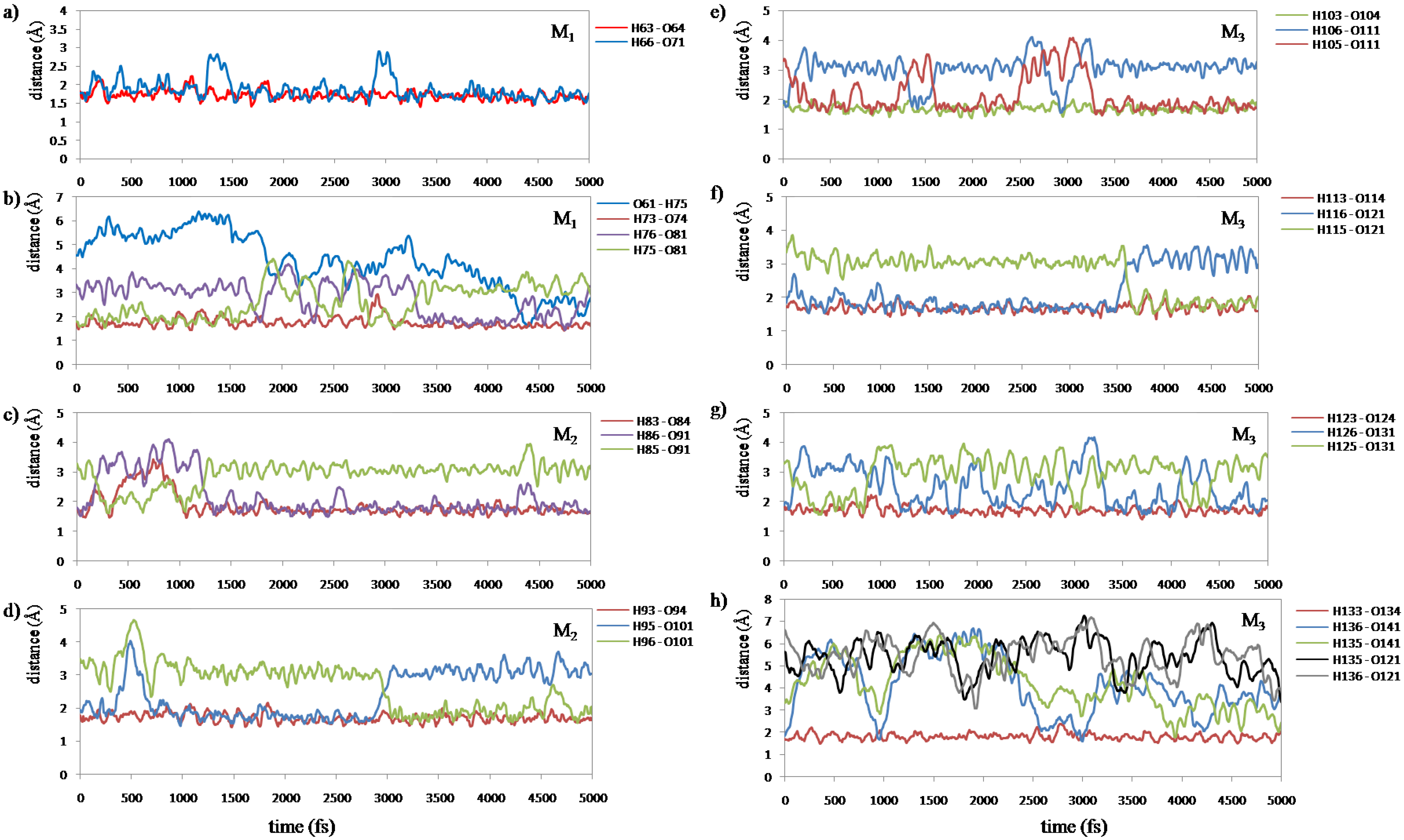
7(C=O···H–O···H–N) type. We measured all of the H-bond lengths during the simulation to examine the configuration change. We assigned these H-bonds into eight groups according to their locations. The changes in the atomic distances of these H-bonds are shown in
Figure 8.
Figure 8a shows that the H-bonds (H63···O64 and H66···O71) near the curve region in the helix are relatively stable throughout the simulation.
Figure 8c–g show that the number of H-bonds is always maintained at two in these areas. In addition, the variation of H-bonds only occurs between O of the backbone’s C=O group and the two H atoms of the water molecule in the vicinity induced by the rotation of the water molecule.
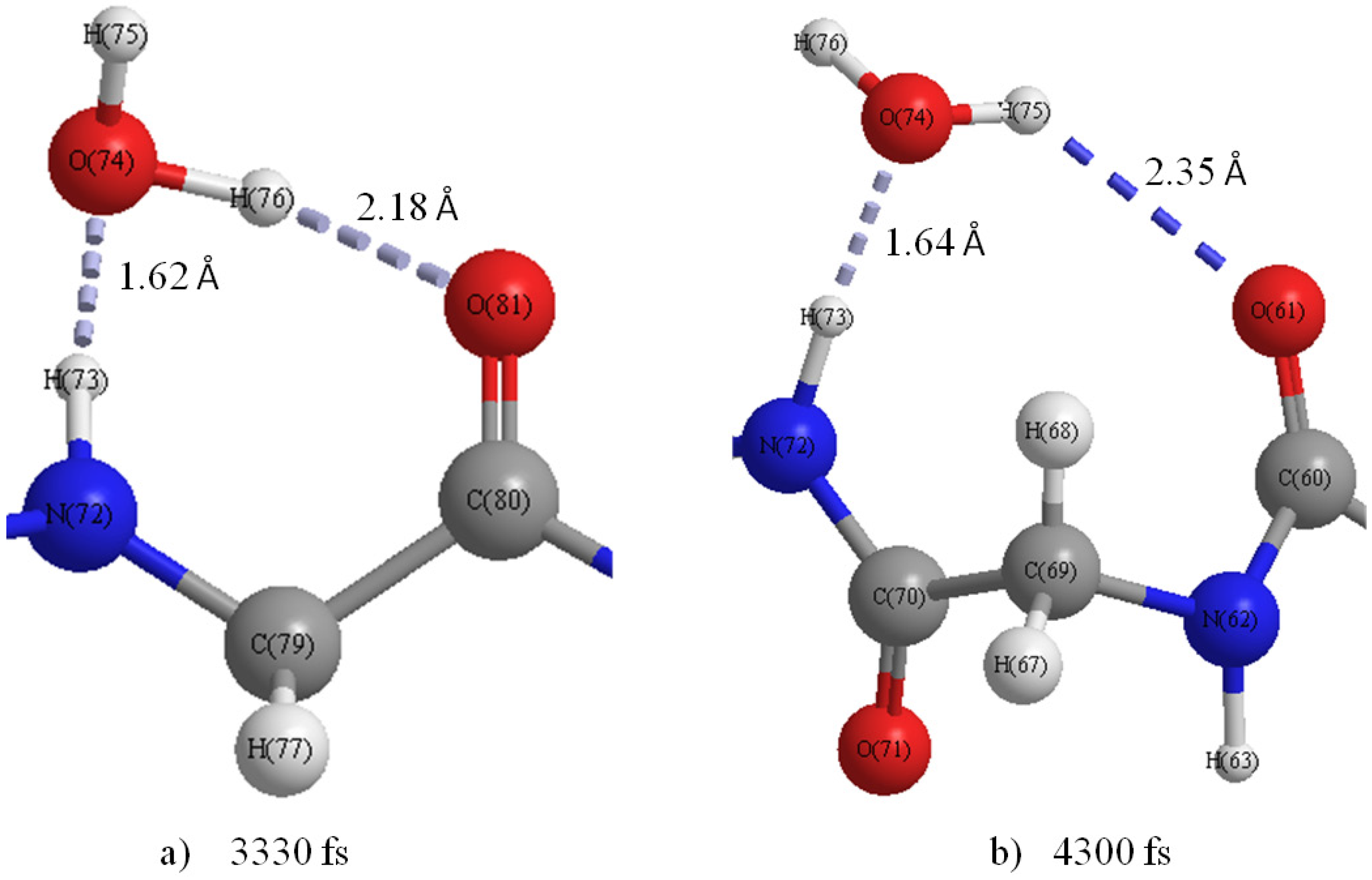
Figure 8b shows that the H-bond of H75···O81 changed to that of H76···O81 at 3330 fs (
Figure 9a), and a new H-bond O61···H75 in C
9(C=O···H–O···H–N) type is observed at 4300 fs (
Figure 9b). Then, it was found that the H-bonds H76···O81 and O61···H75 became stable after 4300 fs. The formation of new C
9(C=O···H–O···H–N) type H-bonds implies that the helix structure of this area becomes more stable and more compact than the initial structure. The variations of H-bonds in the tail region of the peptide are more active than the other regions as shown in
Figure 8h. After 4300 fs, the H135···O141, H135···O121 and H136···O121 distances have a downward trend, but the H136···O141distance increases in contrast. This implies that the H-bond of H136···O141 gradually changes to H135···O141, and it has a tendency to form a new H-bond H136···O121 in the C
9(C=O···H–O···H–N) type as shown in the snapshot at 5000 fs (
Figure 10c). The snapshot structures of interacting space (M
1, M
2 and M
3) and the entire peptide at 5000 fs are shown in
Figure 10a,b, respectively. All the C
7(C=O···H–O···H–N) type H-bonds in the peptide interacting space are stable during the simulation. On the other hand, a new H-bond ring in the C
9(C=O···H–O···H–N) type is formed in front of the interacting space, and the structure of the tail region has the tendency to form another H-bond ring of the C
9(C=O···H–O···H–N) type. The H-bonds in the C
9(C=O···H–O···H–N) type can cause the distortion of the structure leading to the forming of a helix conformation. It can be concluded that the β-strand conformation included in the interacting space (M
3 region) is transformed into a helix conformation due to the newly formed H-bonds.
Figure 8.
Fluctuations of the H-bond distances between peptide and water molecules in the interacting space (5 ps ELG-MD simulation). The numbering of the atoms is shown in
Figure 5. M
1, M
2 and M
3 denote the M
1 region, M
2 region and M
3 region shown in
Figure 5 respectively. More details of the H-bond distances in Panel b and h are shown in
Figure 9 and
Figure 10, respectively.
Figure 8.
Fluctuations of the H-bond distances between peptide and water molecules in the interacting space (5 ps ELG-MD simulation). The numbering of the atoms is shown in
Figure 5. M
1, M
2 and M
3 denote the M
1 region, M
2 region and M
3 region shown in
Figure 5 respectively. More details of the H-bond distances in Panel b and h are shown in
Figure 9 and
Figure 10, respectively.
Figure 9.
Snapshot of H-bond rings in the M1 region at (a) 3330 fs and (b) 4300 fs.
Figure 9.
Snapshot of H-bond rings in the M1 region at (a) 3330 fs and (b) 4300 fs.
Figure 10.
Snapshot at 5000fs of (a) entire peptide (Gly)14. Atoms from 1 to 61 belong to the frozen region; atoms from 62 to 143 belong to the active region; (b) Interacting space (M1, M2 and M3); and (c) H-bond.
Figure 10.
Snapshot at 5000fs of (a) entire peptide (Gly)14. Atoms from 1 to 61 belong to the frozen region; atoms from 62 to 143 belong to the active region; (b) Interacting space (M1, M2 and M3); and (c) H-bond.
It is a big challenge to understand the interactions in biomolecules at the atomic level with the surrounding water molecules because of their transient character and the inhomogeneity of H-bonding in liquid water [
23]. Here, we have proposed a new kind of theoretical investigation on the interactions between peptide and explicit water molecules, and have been able to show the H-bond effects on the conformation transformation of the peptide as a first AIMD application.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









