
Liquid Crystalline Esters of Dibenzophenazines


Abstract
:
1. Introduction
2. Results and Discussion




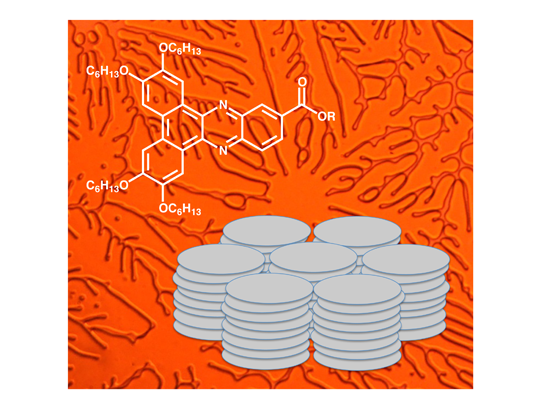{kind=link}
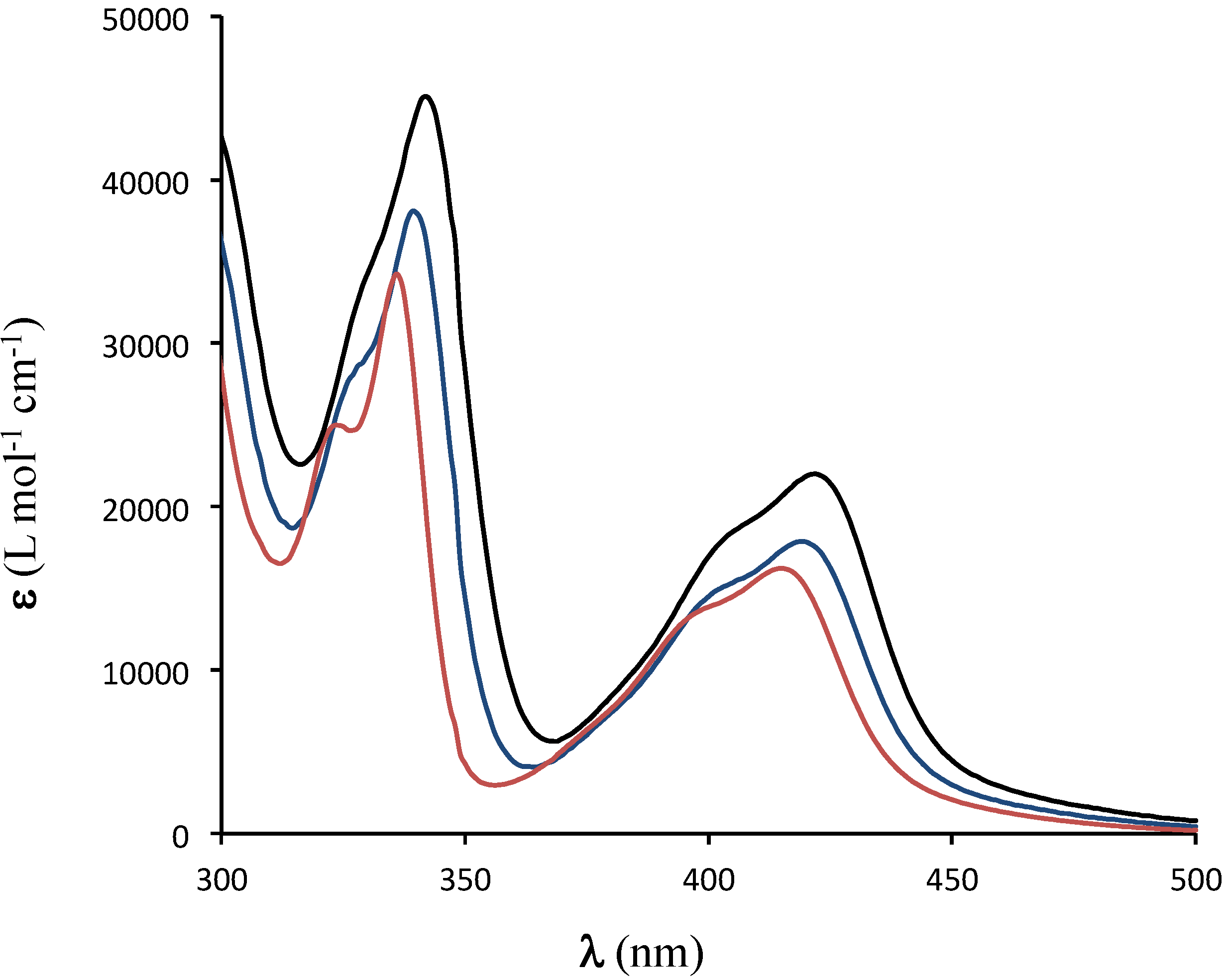{kind=link}
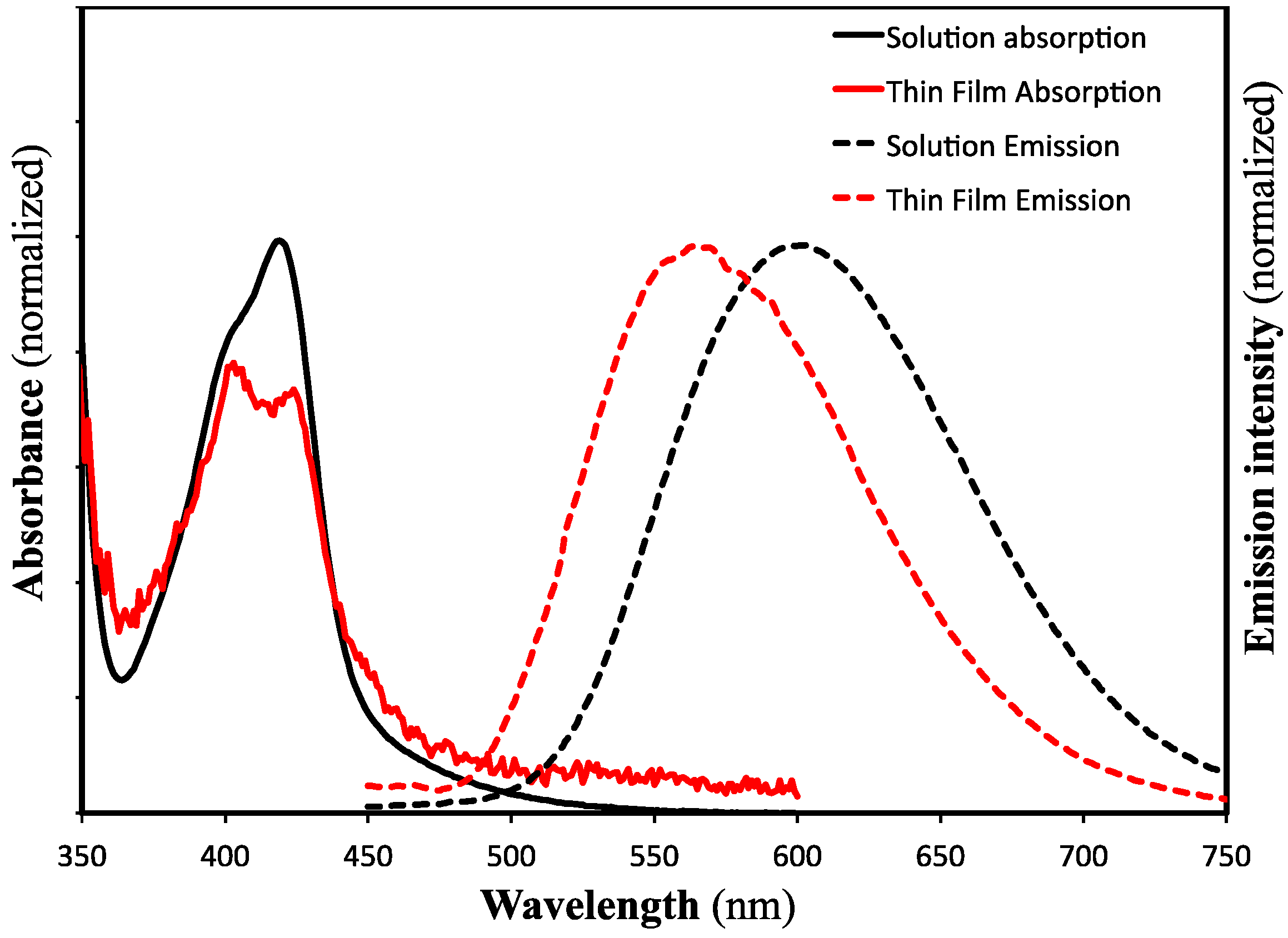{kind=link}
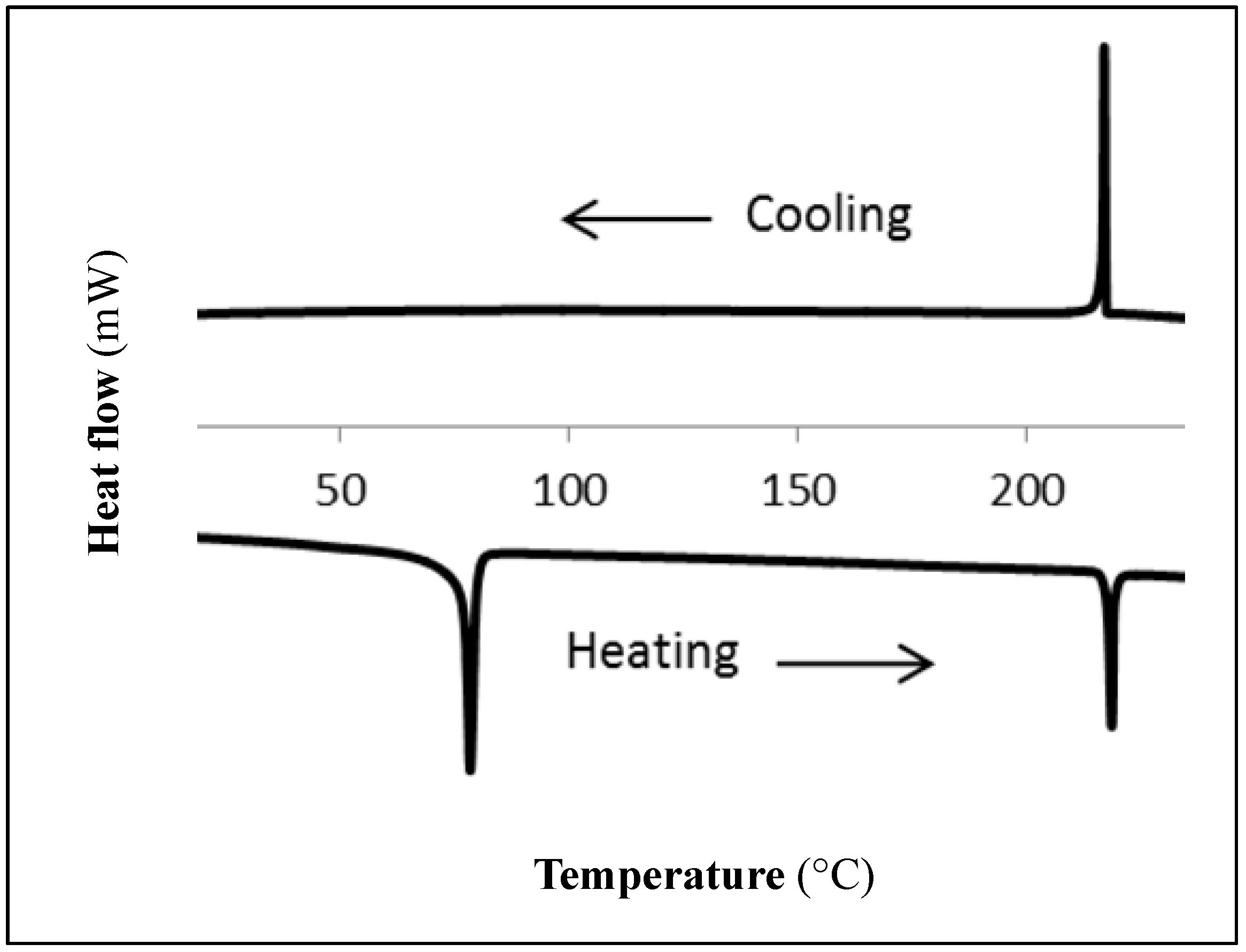{kind=link}
{kind=link}
{kind=link}
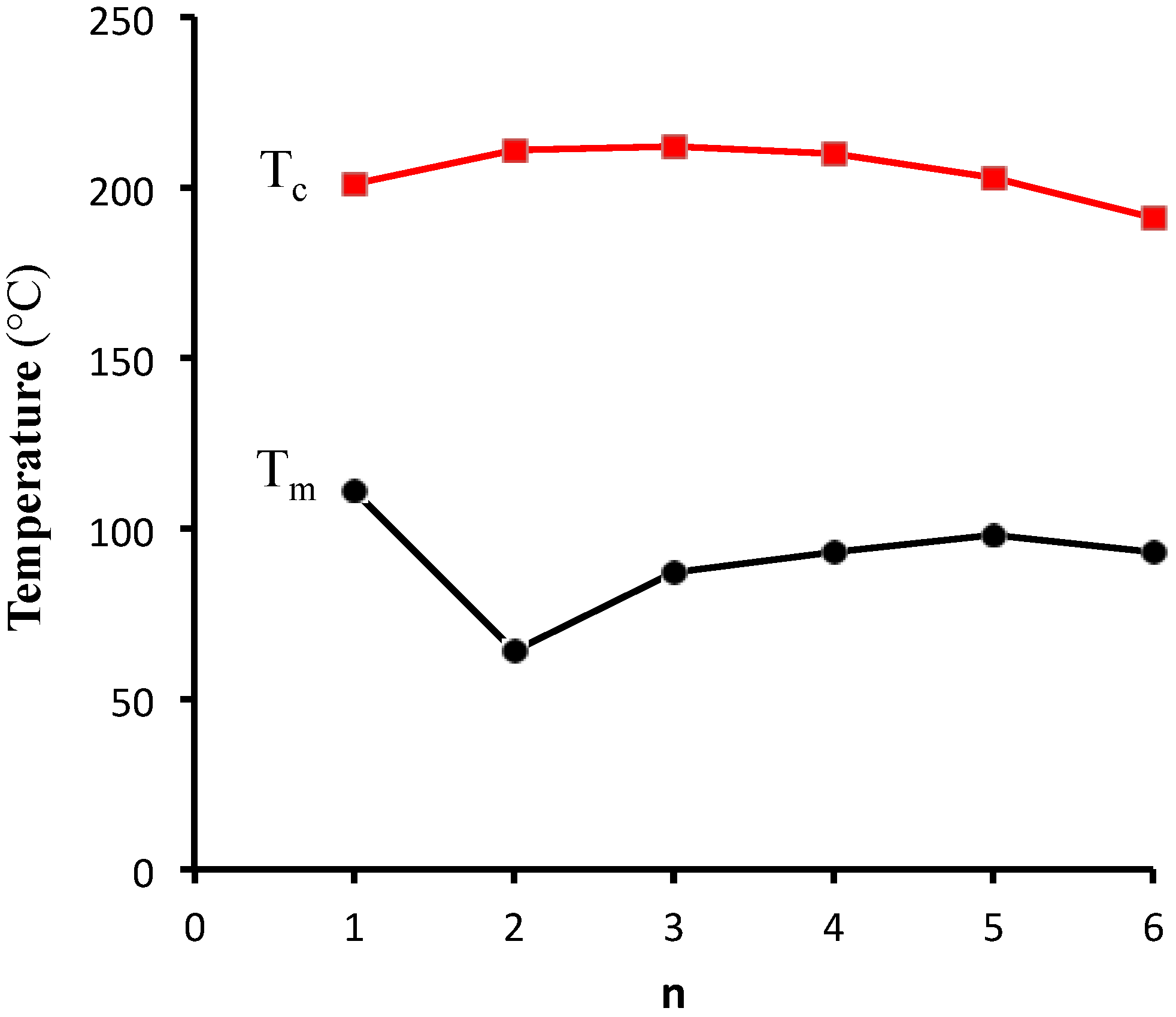{kind=link}
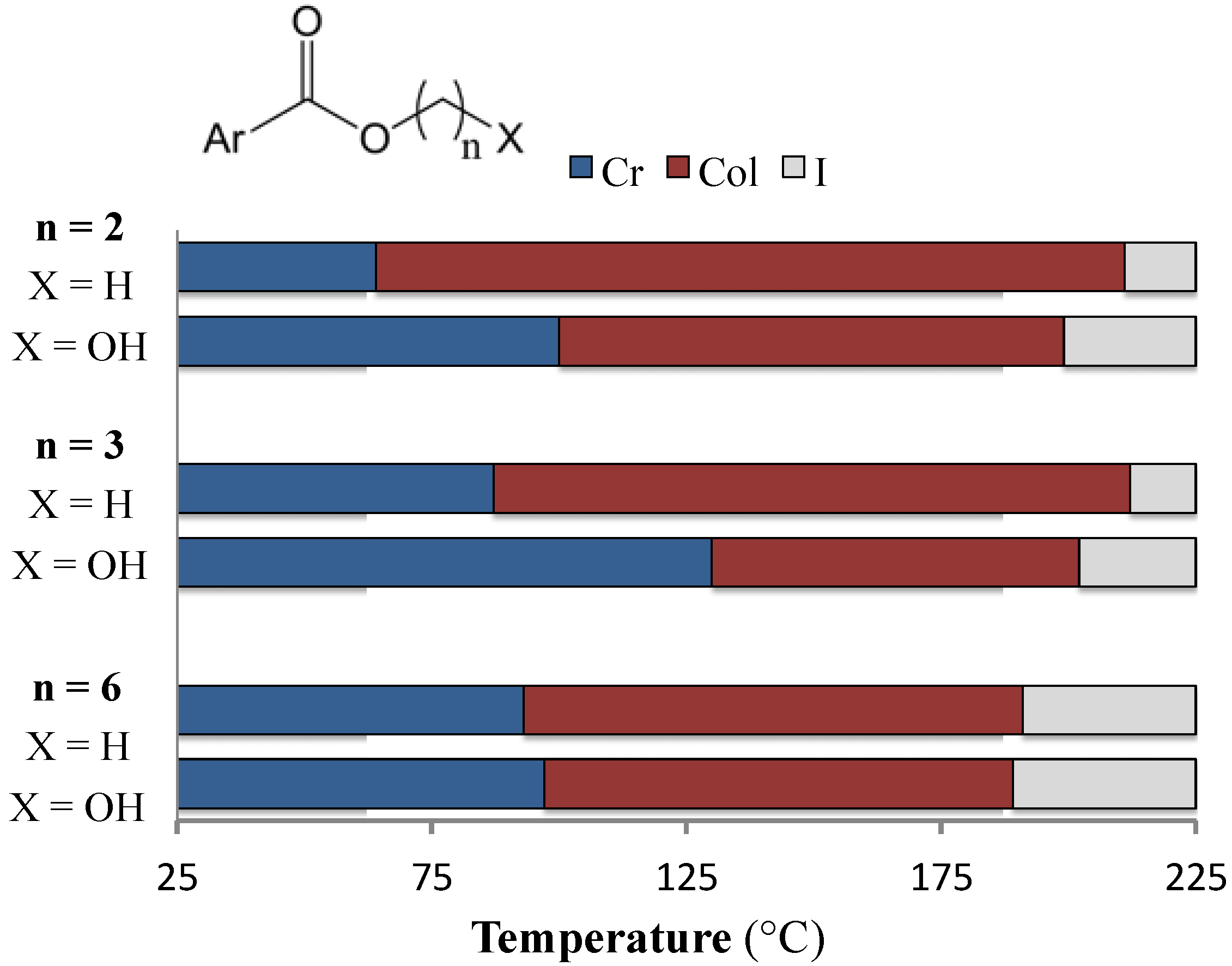{kind=link}
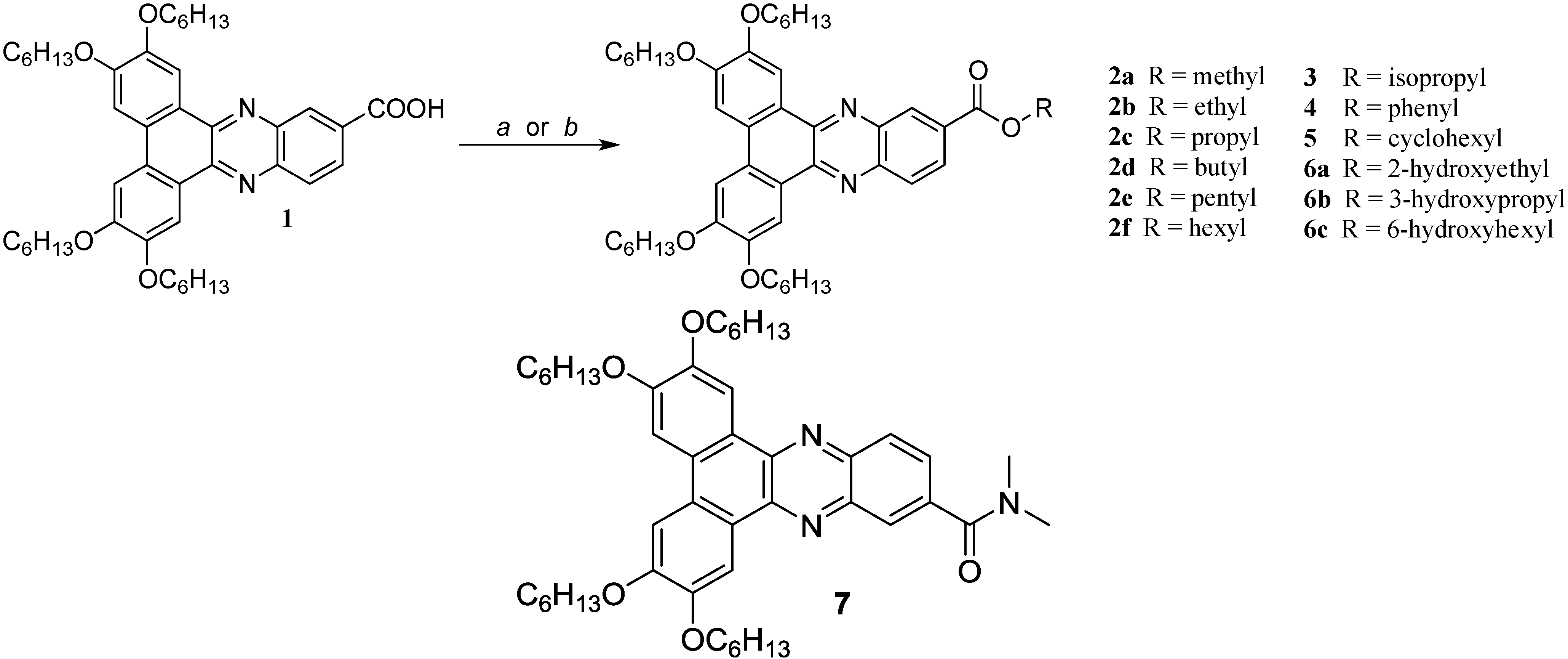{kind=link}
| Compound | Phase | T/°C (ΔH/J g−1) a | Phase | T/°C (ΔH/J g−1) a | Phase b |
|---|---|---|---|---|---|
| 2a c | Cr | 111.3 (39.1) | Colh | 200.6 (4.7) 196.5 (4.8) | I |
| 2b | Cr | 64.3 (45.6) | Colh | 210.8 (8.0) 209.5 (11.1) | I |
| 2c | Cr | 86.8 (40.1) | Colh | 212.1 (7.9) 210.2 (9.8) | I |
| 2d | Cr | 92.5 (42.0) | Colh | 209.6 (8.3) 208.2 (8.6) | I |
| 2e | Cr | 97.5 (68.0) | Colh | 202.9 (7.7) 202.0 (7.7) | I |
| 2f | Cr | 92.7 (61.8) | Colh | 191.3 (4.6) 190.9 (6.3) | I |
| 3 | Cr | 78.6 (32.7) | Colh | 218.8 (9.6) 217.1 (10.6) | I |
| 4 | Cr | 124.7 (38.7) | Colh | 220.5 (4.1) 218.6 (5.8) | I |
| 5 | Cr | 84.7 (18.5) | Colh | 202.5 (5.6) 199.1 (7.3) | I |
| 6a | Cr | 99.7 (15.0) | Colh | 198.7 (6.6) 197.6 (6.5) | I |
| 6b d | Cr | 130.1 (71.8) | Colh | 202.0 (9.4) 201.1 (9.3) | I |
| 6c | Cr | 96.5 (51.2) | Colh | 188.8 (7.9) 187.8 (8.8) | I |
| 7 | Cr | 122.9 (45.5) | Colh | 180.3 (2.9) 180.1 (3.1) | I |
| Compound | Temperature (°C) | d-spacings (Å) | Miller Index (hkl) | Phase (Lattice Constants) |
|---|---|---|---|---|
| 2a | 150 | 17.3 | 100 | Colh |
| 4.3 | Alkyl Halo | (a = 20.0 Å) | ||
| 3.5 | π–π stacking | – | ||
| 2b | 179 | 17.7 | 100 | Colh |
| 5 | Alkyl Halo | (a = 20.5 Å) | ||
| 3.8 | π–π stacking | – | ||
| 2c | 177 | 17.4 | 100 | Colh |
| 4.9 | Alkyl Halo | (a = 20.1 Å) | ||
| 2d | 178 | 17.7 | 100 | Colh |
| 4.9 | Alkyl Halo | (a = 20.5 Å) | ||
| 2e | 179 | 17.9 | 100 | Colh |
| 4.7 | Alkyl Halo | (a = 20.7 Å) | ||
| 3.6 | π–π stacking | – | ||
| 2f | 155 | 18.5 | 100 | Colh |
| 4.6 | Alkyl Halo | (a = 21.4 Å) | ||
| 3 | 185 | 17.7 | 100 | Colh |
| 10.3 | 110 | (a = 20.4 Å) | ||
| 5.4 | Alkyl Halo | – | ||
| 4 | 175 | 17.7 | 100 | Colh |
| 4.5 | Alkyl Halo | (a = 20.4 Å) | ||
| 5 | 155 | 18.1 | 100 | Colh |
| 10.6 | 110 | (a = 20.9 Å) | ||
| 6.6 | Alkyl Halo | – | ||
| 6a | 160 | 17.3 | 100 | Colh |
| 5.1 | Alkyl Halo | (a = 20.0 Å) | ||
| 3.6 | π–π stacking | – | ||
| 6b | 170 | 17.5 | 100 | Colh |
| 4.6 | Alkyl Halo | (a = 20.2 Å) | ||
| 3.5 | π–π stacking | – | ||
| 6c | 157 | 18.3 | 100 | Colh |
| 5.1 | Alkyl Halo | (a = 21.1 Å) | ||
| 7 | 169 | 17.1 | 100 | Colh |
| 4.7 | Alkyl Halo | (a = 19.8 Å) |





3. Experimental Section
3.1. Synthesis Method 1 (Oxalyl Chloride Method)
3.2. Synthesis Method 2 (EDCI/DMAP Method)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaafarani, B.R. Discotic liquid crystals for opto-electronic applications. Chem. Mater. 2011, 23, 378–396. [Google Scholar] [CrossRef]
- Laschat, S.; Baro, A.; Steinke, N.; Giesselmann, F.; Hägele, C.; Scalia, G.; Judele, R.; Kapatsina, E.; Sauer, S.; Schreivogel, A.; et al. Discotic liquid crystals: From tailor-made synthesis to plastic electronics. Angew. Chem. Int. Ed. 2007, 46, 4832–4887. [Google Scholar] [CrossRef]
- Imrie, C.T.; Henderson, P.A. Liquid crystal dimers and higher oligomers: Between monomers and polymers. Chem. Soc. Rev. 2007, 36, 2096–2124. [Google Scholar] [CrossRef] [PubMed]
- Bushby, R.J.; Kawata, K. Liquid crystals that affected the world: Discotic liquid crystals. Liq. Cryst. 2011, 38, 1415–1426. [Google Scholar] [CrossRef]
- Kumar, S. Self-organization of disc-like molecules: Chemical aspects. Chem. Soc. Rev. 2006, 35, 83–109. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Triphenylene-based discotic liquid crystal dimers, oligomers and polymers. Liq. Cryst. 2005, 32, 1089–1113. [Google Scholar] [CrossRef]
- Keuker-Baumann, S.; Bock, H.; Sala, F.D.; Benning, S.A.; Haßheider, T.; Frauenheim, T.; Kitzerow, H.-S. Absorption and luminescence spectra of electroluminescent liquid crystals with triphenylene, pyrene and perylene units. Liq. Cryst. 2001, 28, 1105–1113. [Google Scholar] [CrossRef]
- Hassheider, T.; Benning, S.A.; Kitzerow, H.-S.; Achard, M.-F.; Bock, H. Color-tuned electroluminescence from columnar liquid crystalline alkyl arenecarboxylates. Angew. Chem. Int. Ed. 2001, 40, 2060–2063. [Google Scholar] [CrossRef]
- Benning, S.; Kitzerow, H.-S.; Bock, H.; Achard, M.-F. Fluorescent columnar liquid crystalline 3,4,9,10-tetra-(n-alkoxycarbonyl)-perylenes. Liq. Cryst. 2000, 27, 901–906. [Google Scholar] [CrossRef]
- Ito, S.; Wehmeier, M.; Brand, J.D.; Kübel, C.; Epsch, R.; Rabe, J.P.; Müllen, K. Synthesis and self-assembly of functionalized hexa-peri-hexabenzocoronenes. Chem. Eur. J. 2000, 6, 4327–4342. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.J.; Lavigueur, C.; Ke, Y.-C.; Williams, V.E. Self-assembly of hydrogen-bonded molecules: Discotic and elliptical mesogens. J. Mater. Chem. 2005, 15, 4062–4068. [Google Scholar] [CrossRef]
- Bozek, K.J.A.; Williams, V.E. Folded discotic dimers. Soft Matter 2014, 10, 5749–5754. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitani, D.; Lien, E.J. Aromatic substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Szydłowska, J.; Krzyczkowska, P.; Salamończyk, M.; Górecka, E.; Pociecha, D.; Maranowski, B.; Krówczyński, A. Gelling and fluorescent mesogens of quinoxaline analogs. J. Mater. Chem. C 2013, 1, 6883–6889. [Google Scholar] [CrossRef]
- Ong, C.W.; Hwang, J.-Y.; Tzeng, M.-C.; Liao, S.-C.; Hsu, H.-F.; Chang, T.-H. Dibenzo[a,c]phenazine with six-long alkoxy chains to probe optimization of mesogenic behavior. J. Mater. Chem. 2007, 17, 1785–1790. [Google Scholar] [CrossRef]
- Ong, C.W.; Hwang, C.-Y.; Liao, S.-C.; Pan, C.-H.; Chang, T.-H. Indent and bulge-discogens for controlling the columnar mesophase. J. Mater. Chem. 2009, 19, 5149–5154. [Google Scholar] [CrossRef]
- Kumar, S. Chemistry of Discotic Liquid Crystals: From Monomers to Polymers; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Lavigueur, C.; Foster, E.J.; Williams, V.E. Self-assembly of discotic mesogens in solution and in liquid crystalline phases: Effects of substituent position and hydrogen bonding. J. Am. Chem. Soc. 2008, 130, 11791–11800. [Google Scholar] [CrossRef] [PubMed]
- Collard, D.M.; Lillya, C.P. Control of thermal phase behavior of disklike molecules by modification of side-chain structure. J. Am. Chem. Soc. 1989, 111, 1829–1830. [Google Scholar] [CrossRef]
- Collard, D.M.; Lillya, C.P. Structure–property relationships for the thermal phase behavior of discotic liquid crystals: The effect of branching and unsaturation in the side chains of disklike molecules. J. Am. Chem. Soc. 1991, 113, 8577–8583. [Google Scholar] [CrossRef]
- Varshney, S.K.; Prasad, V.; Takezoe, H. Room-temperature discotic cholesteric and nematic phases: influence of 3,7-dimethyloctane peripheral chain on the molecular self-assembly of radial polyalkynylbenzene. Liq. Cryst. 2011, 38, 53–60. [Google Scholar] [CrossRef]
- Stackhouse, P.J.; Hird, M. Influence of branched chains on the mesomorphic properties of symmetrical and unsymmetrical triphenylene discotic liquid crystals. Liq. Cryst. 2008, 35, 597–607. [Google Scholar] [CrossRef]
- Paleos, C.M.; Tsiourvas, D. Supramolecular hydrogen-bonded liquid crystals. Liq. Cryst. 2001, 28, 1127–1161. [Google Scholar] [CrossRef]
- Wang, B.Q.; Zhao, K.Q.; Hu, P.; Yu, W.H.; Gao, C.Y.; Shimizu, Y. Tuning hydrogen-bonding with amide groups for stable columnar mesophases of triphenylene discogens. Mol. Cryst. Liq. Cryst. 2007, 479, 135/[1173]–150/[1188]. [Google Scholar] [CrossRef]
- Boden, N.; Bushby, R.J.; Lu, Z.B. A rational synthesis of polyacrylates with discogenic side groups. Liq. Cryst. 1998, 25, 47–58. [Google Scholar] [CrossRef]
- Grafe, A.; Janietz, D.; Frese, T.; Wendorff, J.H. Star-shaped discotic oligomesogens based on radial pentakisphenylethynylbenzene moieties. Chem. Mater. 2005, 17, 4979–4984. [Google Scholar] [CrossRef]
- Foster, E.J.; Jones, R.B.; Lavigueur, C.; Williams, V.E. Structural factors controlling the self-assembly of columnar liquid crystals. J. Am. Chem. Soc. 2006, 128, 8569–8574. [Google Scholar] [CrossRef] [PubMed]
- Lavigueur, C.; Foster, E.J.; Williams, V.E. A simple and inexpensive capillary furnace for variable-temperature X-ray diffraction. J. Appl. Crystallogr. 2008, 41, 214–216. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozek, K.J.A.; Ho, K.I.; Saint-Martin, T.; Argyropoulos, P.; Williams, V.E. Liquid Crystalline Esters of Dibenzophenazines. Materials 2015, 8, 270-284. https://doi.org/10.3390/ma8010270
Bozek KJA, Ho KI, Saint-Martin T, Argyropoulos P, Williams VE. Liquid Crystalline Esters of Dibenzophenazines. Materials. 2015; 8(1):270-284. https://doi.org/10.3390/ma8010270
Chicago/Turabian StyleBozek, Kevin John Anthony, Kuan I Ho, Tom Saint-Martin, Panos Argyropoulos, and Vance E. Williams. 2015. "Liquid Crystalline Esters of Dibenzophenazines" Materials 8, no. 1: 270-284. https://doi.org/10.3390/ma8010270
APA StyleBozek, K. J. A., Ho, K. I., Saint-Martin, T., Argyropoulos, P., & Williams, V. E. (2015). Liquid Crystalline Esters of Dibenzophenazines. Materials, 8(1), 270-284. https://doi.org/10.3390/ma8010270