
First-Principles Investigation of Phase Stability, Electronic Structure and Optical Properties of MgZnO Monolayer



Abstract
:1. Introduction
2. Calculational Methods
3. Results and Discussion
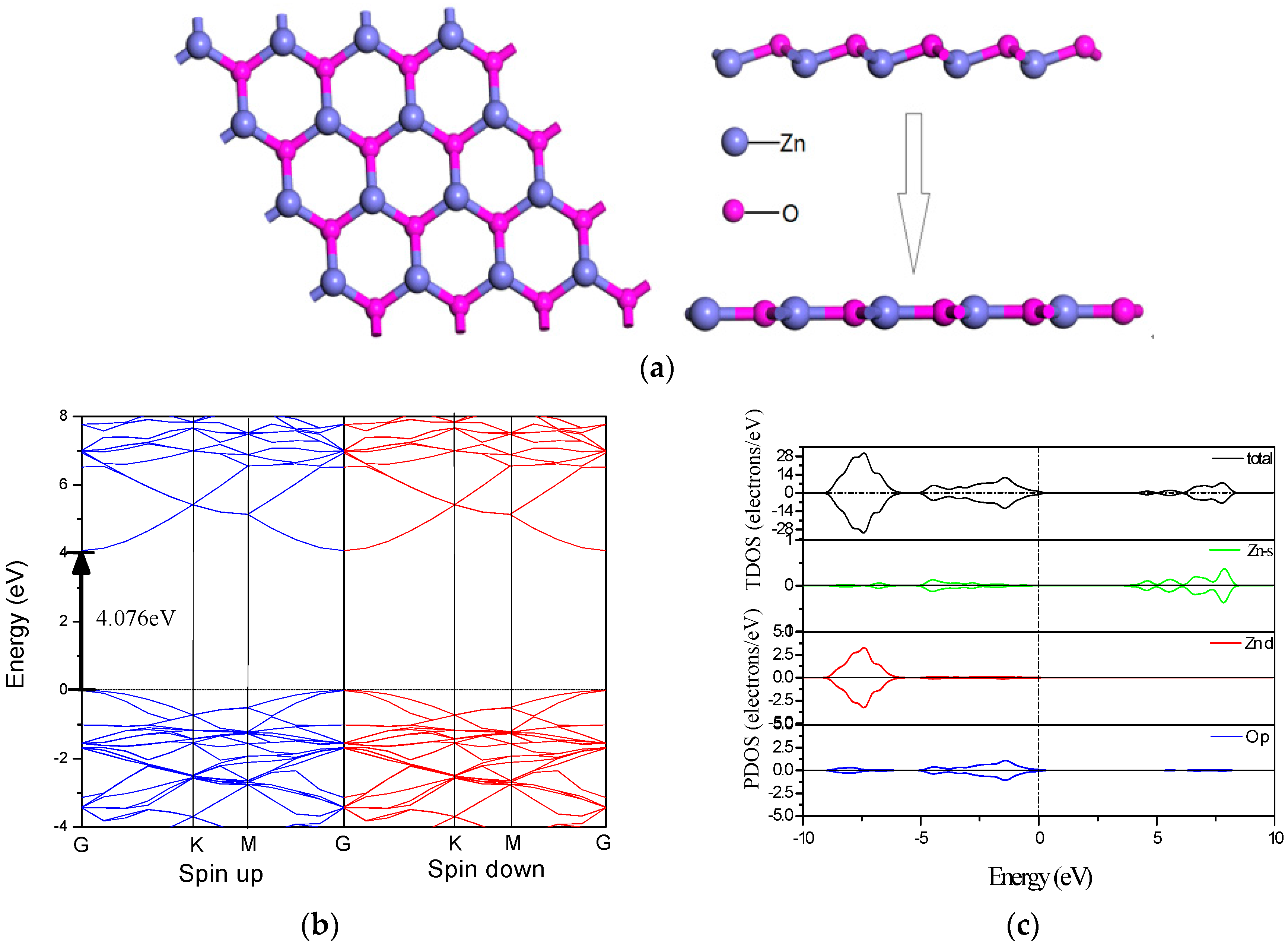
3.1. The Pristine ZnO Monolayer
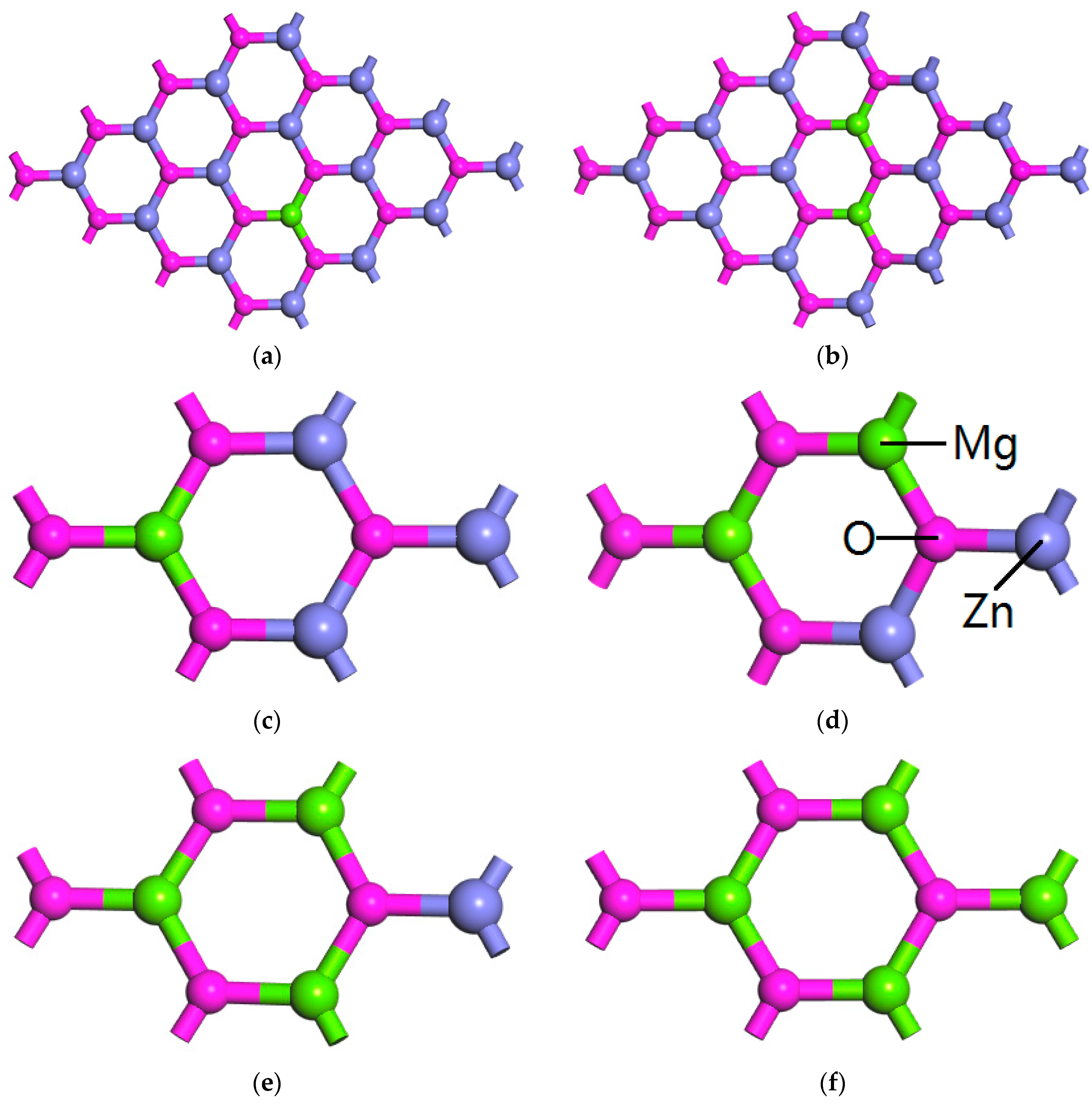
3.2. Phase Stability of MgZnO Monolayer
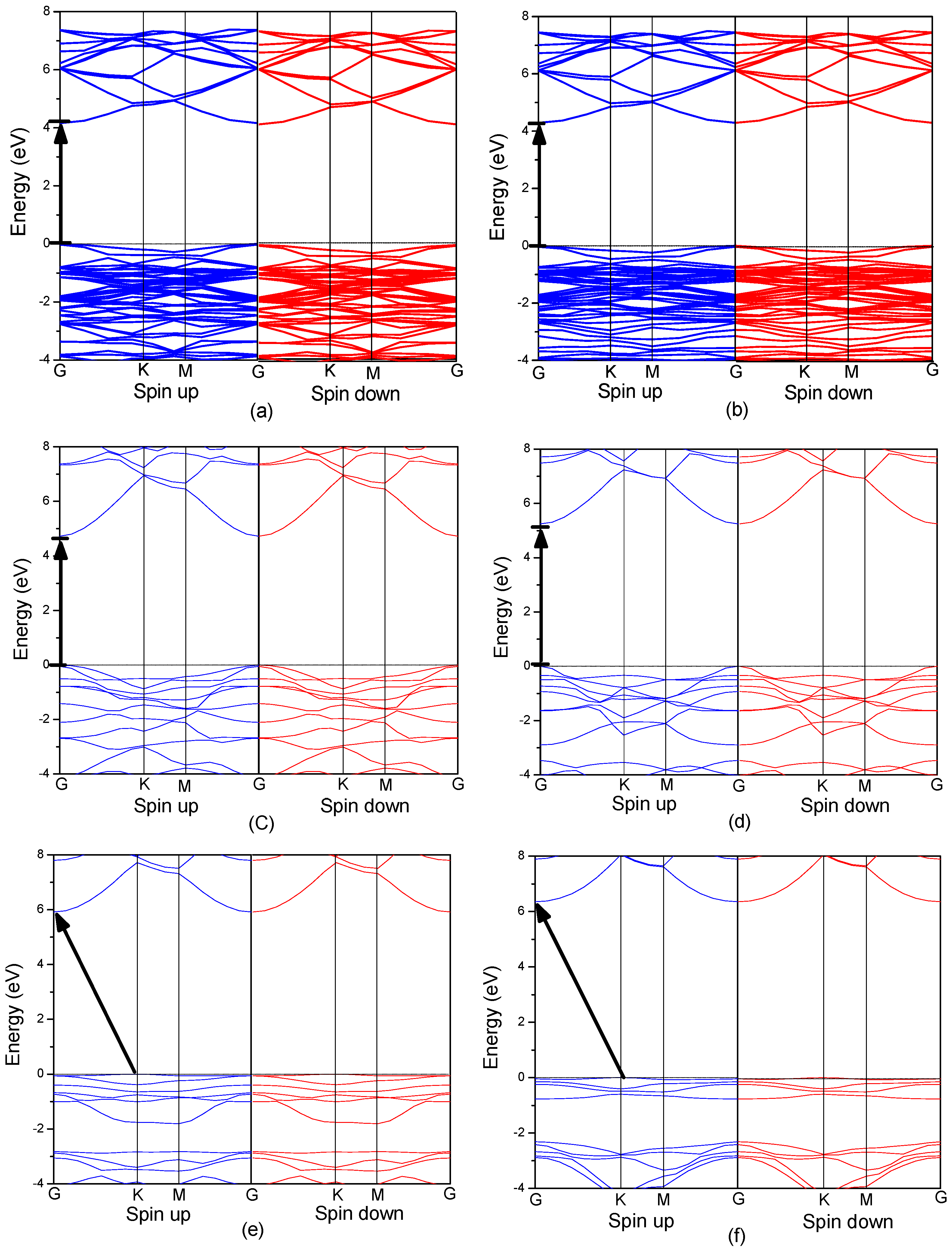
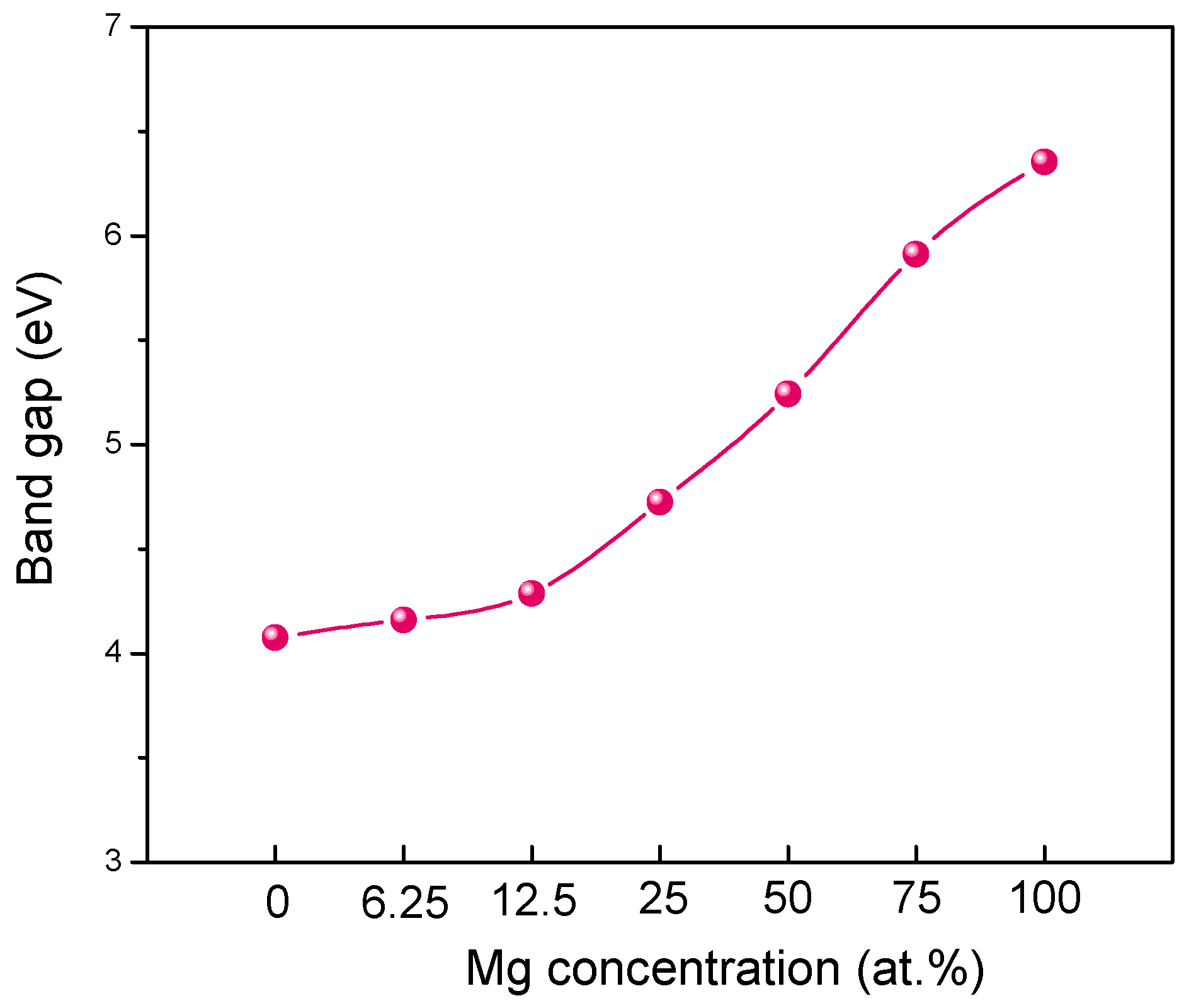
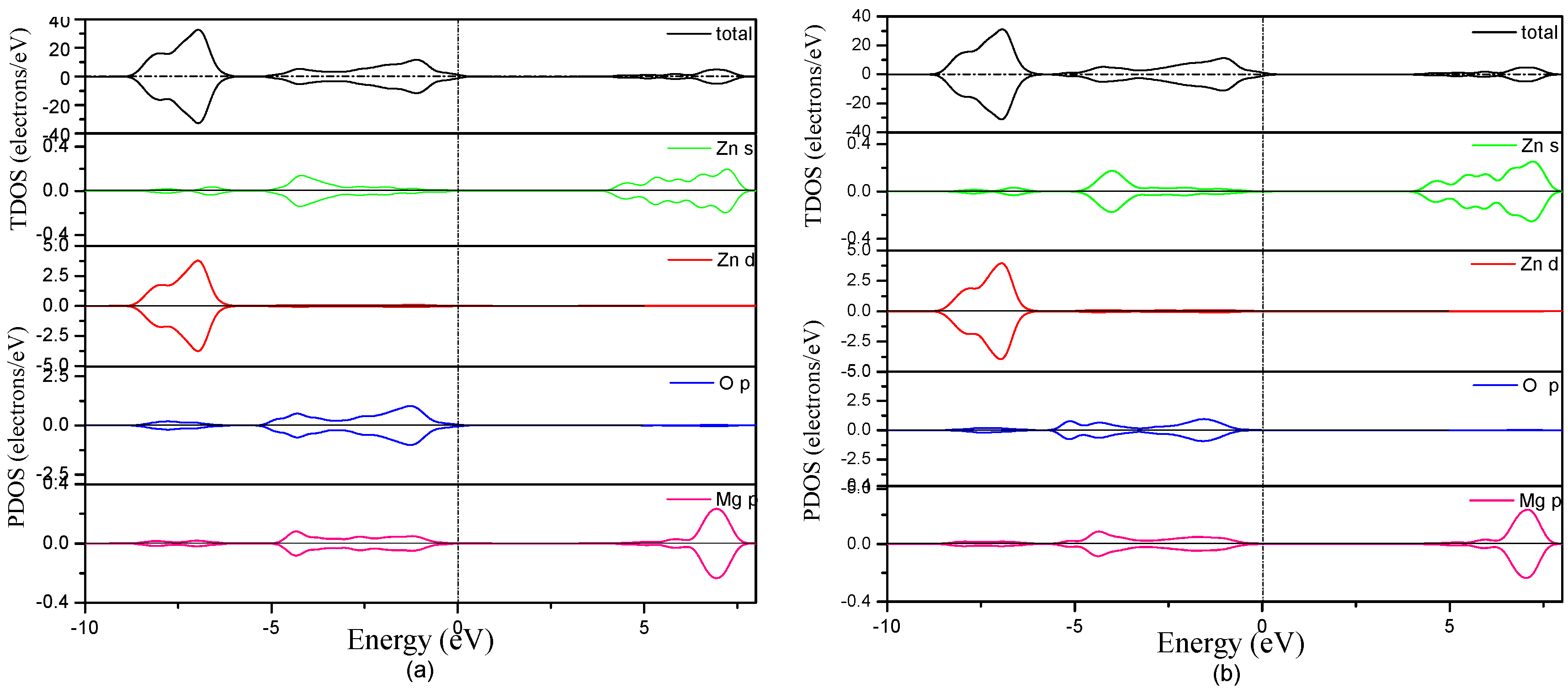
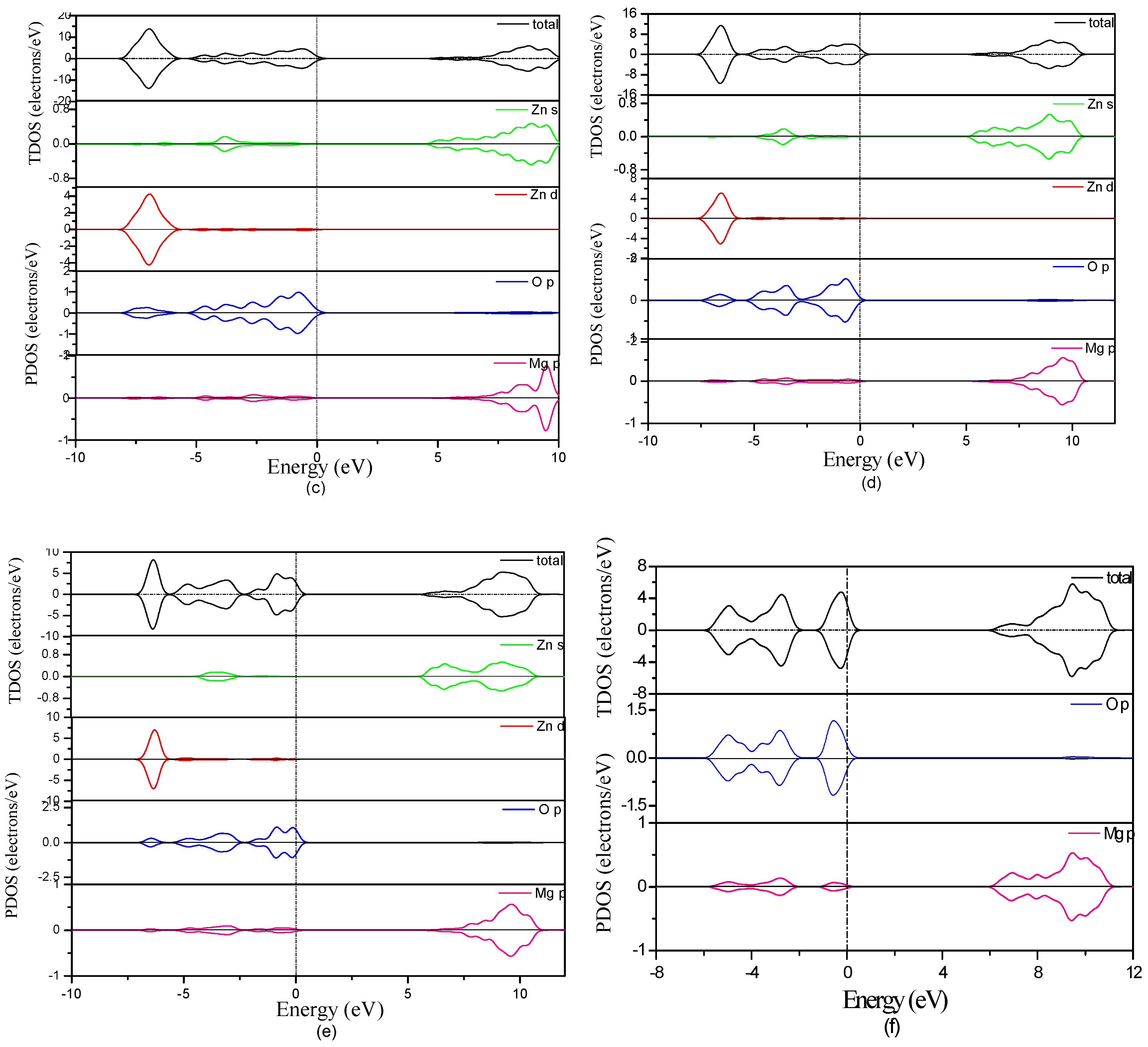
3.3. Electronic Structure of MgZnO Monolayer
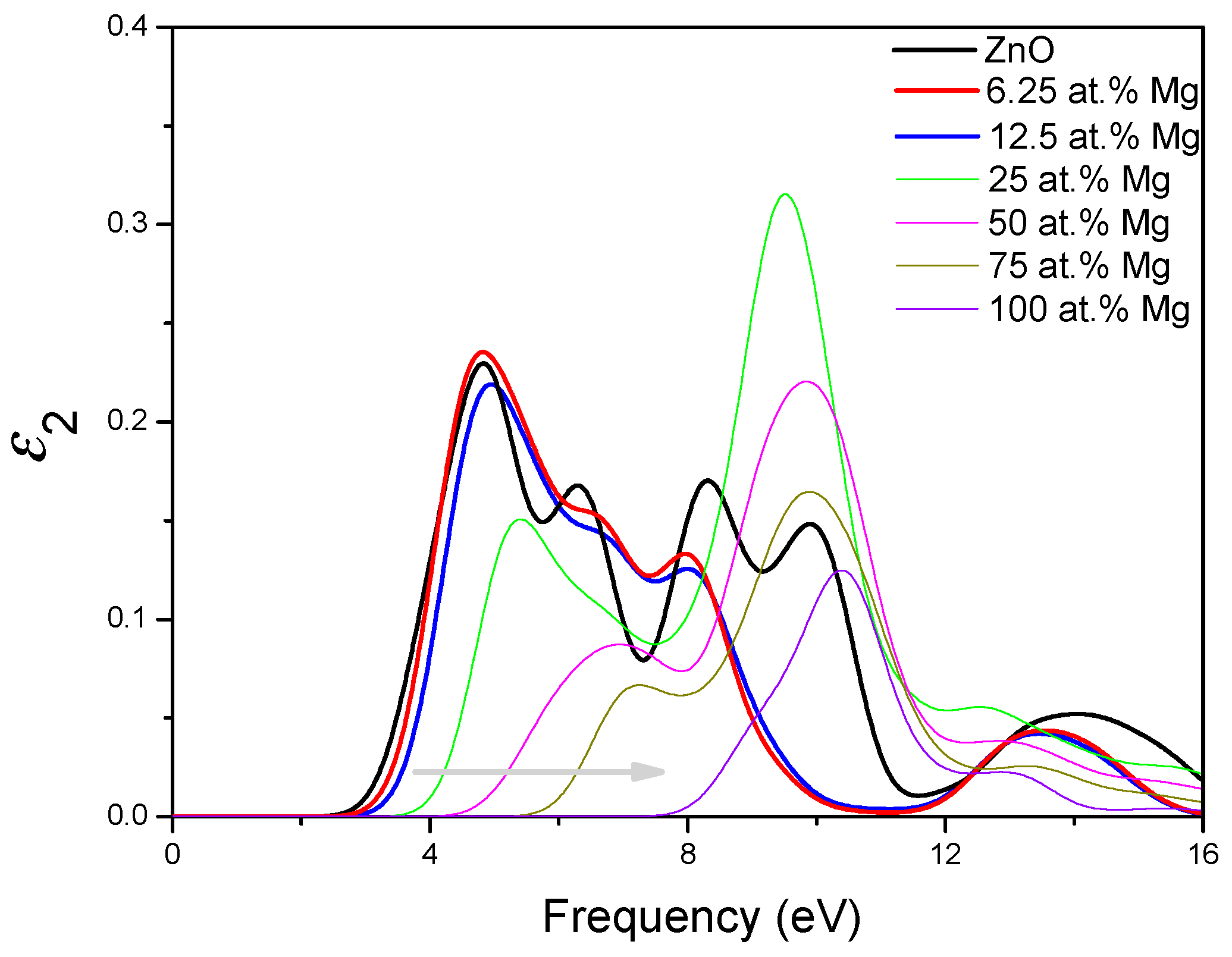
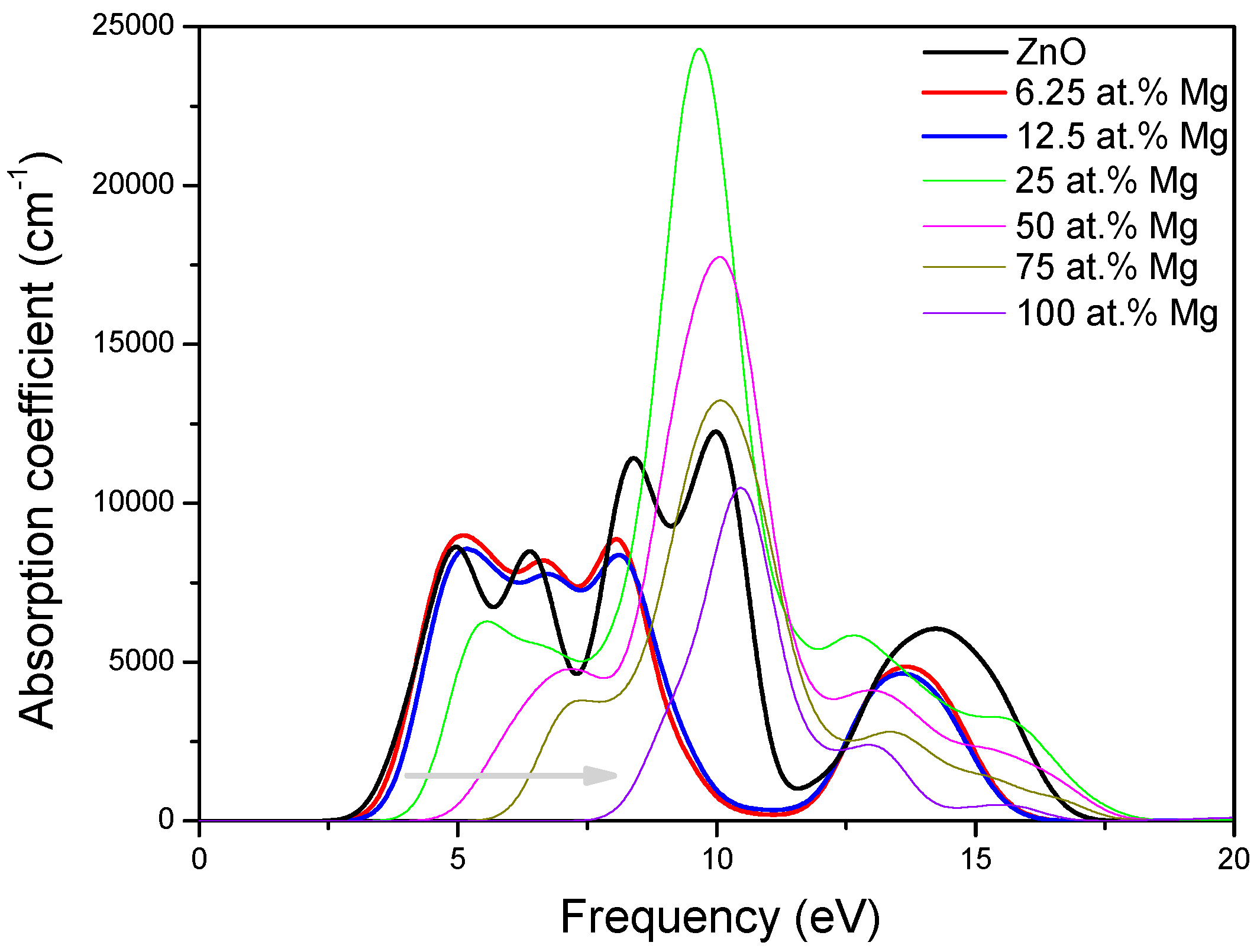
3.4. Optical Properties of MgZnO Monolayer
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xu, M.S.; Liang, T.; Shi, M.M.; Chen, H.Z. Graphene-like two-dimensional materials. Chem. Rev. 2013, 113, 3766–3798. [Google Scholar] [CrossRef] [PubMed]
- Ozgur, U.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Dogan, S.; Avrutin, V.; Cho, S.J.; Morkoc, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 2005, 98, 041301. [Google Scholar] [CrossRef]
- Law, M.; Greene, L.E.; Johnson, J.C.; Saykally, R.; Yang, P.D. Nanowire dye-sensitized solar cells. Nat. Mater. 2005, 4, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Mende, L.; MacManus-Driscoll, J.L. ZnO-nanostructures, defects, and devices. Mater. Today 2007, 10, 40–48. [Google Scholar] [CrossRef]
- Wang, Z.L. ZnO nanowire and nanobelt platform for nanotechnology. Mater. Sci. Eng. R 2009, 64, 33–71. [Google Scholar] [CrossRef]
- Freeman, C.L.; Claeyssens, F.; Allan, N.L. Graphitic nanofilms as precursors to wurtzite films: Theory. Phys. Rev. Lett. 2006, 96, 066102. [Google Scholar] [CrossRef] [PubMed]
- Tusche, C.; Meyerheim, H.L.; Kirschner, J. Observation of depolarized ZnO(0001) monolayers: Formation of unreconstructed planar sheets. Phys. Rev. Lett. 2007, 99, 026102. [Google Scholar] [CrossRef] [PubMed]
- Weirum, G.; Barcaro, G.; Fortunelli, A.; Weber, F.; Schennach, R.; Surnev, S.; Netzer, F.P. Growth and Surface Structure of Zinc Oxide Layers on a Pd(111) Surface. J. Phys. Chem. C 2010, 114, 15432–15439. [Google Scholar] [CrossRef]
- Quang, H.T.; Bachmatiuk, A.; Dianat, A.; Ortmann, F.; Zhao, J.; Warner, J.H.; Eckert, J.; Cunniberti, G.; Rummeli, M.H. In situ observations of free-standing graphene-like mono- and bilayer ZnO membranes. ACS Nano 2015, 9, 11408–11413. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sorescu, D.C.; Deng, X.Y. Tunable lattice constant and band gap of single- and few-layer ZnO. J. Phys. Chem. Lett. 2016, 7, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Nayak, S.K.; Chelliah, P.; Raht, M.K.; Parida, B. Observations of two-dimensional monolayer zinc oxide. Mater. Res. Bull. 2016, 75, 134–138. [Google Scholar] [CrossRef]
- Schmidt, T.M.; MiWa, R.H.; Fazzio, A. Ferromagnetic coupling in a Co-doped graphenelike ZnO sheet. Phys. Rev. B 2010, 81, 195413. [Google Scholar] [CrossRef]
- Guo, H.Y.; Zhao, Y.; Lu, N.; Kang, E.J.; Zeng, X.C.; Wu, X.J.; Yang, J.L. Tunable Magnetism in a Nonmetal-Substituted ZnO Monolayer: A First-Principles Study. J. Phys. Chem. C 2012, 116, 11336–11342. [Google Scholar] [CrossRef]
- Tu, Z.C. First-principles study on physical properties of a single ZnO monolayer with graphene-like structure. J. Comput. Theor. Nanosci. 2010, 7, 1182–1186. [Google Scholar] [CrossRef]
- He, A.L.; Wang, X.Q.; Lu, Y.H.; Feng, Y.P. Adsorption of an Mn atom on a ZnO sheet and nanotube: A density functional theory study. J. Phys. Condens. Matter 2010, 22, 175501. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Sun, D.; Xu, D.S.; Tian, X.H.; Huang, Y.W. Tuning electronic structure and optical properties of ZnO monolayer by Cd doping. Ceram. Int. 2016, 42, 10997–11002. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.Q.; Kong, J.H.; Yu, M.H.; Jin, H.Y. Syntheis of Mg-doped hierarchical ZnO nanostructures via hydrothermal method and their optical properties. J. Alloys Compd. 2016, 657, 261–267. [Google Scholar] [CrossRef]
- Schleife, A.; Eisenacher, M.; Rödl, C.; Fuchs, F.; Furthmüller, J.; Bechstedt, F. S Ab initio description of heterostructural alloys: Thermodynamic and structural properties of MgxZn1−xO and CdxZn1−xO. Phys. Rev. B 2010, 81, 245210. [Google Scholar] [CrossRef]
- Kasahara, Y.; Oshima, Y.; Falson, J.; Kozuka, Y.; Tsukazaki, A.; Kawasaki, M.; Iwasa, Y. Correlation-Enhanced Effective Mass of Two-Dimensional Electrons in MgxZn1−xO/ZnO Heterostructures. Phys. Rev. Lett. 2012, 109, 246401. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Takahashi, N.; Nakagawa, Y.; Hiramatsu, T.; Kato, H. Nonlinear characteristics of structural properties and spontaneous polarization in wurtzite MgxZn1−xO: A first-principles study. Phys. Rev. B 2013, 88, 075203. [Google Scholar] [CrossRef]
- Ni, P.N.; Shan, C.X.; Li, B.H.; Shen, D.Z. High Mg-content wurtzite MgZnO alloys and their application in deep-ultraviolet light-emitters pumped by accelerated electrons. Appl. Phys. Lett. 2014, 104, 032107. [Google Scholar] [CrossRef]
- Fan, X.F.; Sun, H.D.; Shen, Z.X.; Kuo, J.L.; Lu, Y.M. A first-principle analysis on the phase stabilities, chemical bonds and band gaps of wurtzite structure AxZn1−xO alloys (A = Ca, Cd, Mg). J. Phys. Condens. Matter 2008, 20, 235221. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Z.; Kulinich, S.A.; Yan, J.; Li, Z.G.; He, J.P.; Kan, C.X.; Zeng, H.B. Epitaxial ZnO nanowire-on-nanoplate structures as efficient and transferable field emitters. Adv. Mater. 2013, 25, 5750. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, A.; Tamura, K.; Kawasaki, M.; Makino, T.; Segawa, Y.; Tang, Z.K.; Wong, G.K.L.; Matsumoto, Y.; Koinuma, H. Room-temperature stimulated emission of excitons in ZnO/(Mg, Zn)O superlattices. Appl. Phys. Lett. 2000, 77, 2204. [Google Scholar] [CrossRef]
- Makino, T.; Segawa, Y.; Kawasaki, M.; Koinuma, H. Optical properties of excitons in ZnO-based quantum well heterostructures. Semicond. Sci. Technol. 2005, 20, S78. [Google Scholar] [CrossRef]
- Sun, J.W.; Zhang, B.P. Well-width dependence of exciton-longitudinal-optical- phonon coupling in MgZnO/ZnO single quantum wells. Nanotechnology 2008, 19, 485401. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.Q.; Zhu, L.P.; Ye, Z.Z.; He, H.P.; Zhang, Y.Z.; Huang, F.; Qiu, M.X.; Zeng, Y.J.; Liu, F.; Jaeger, W. Room-temperature photoluminescence from ZnO/ZnMgOZnO/ZnMgO multiple quantum wells grown on Si(111) substrates. Appl. Phys. Lett. 2007, 91, 022103. [Google Scholar] [CrossRef]
- Makino, T.; Segawa, Y.; Kawasaki, M.; Ohtomo, A.; Shiroki, R.; Tamura, K.; Yasuda, T.; Koinuma, H. Band gap engineering based on MgxZn1−xO and CdyZn1−yO ternary alloy films. Appl. Phys. Lett. 2001, 78, 1237–1240. [Google Scholar] [CrossRef]
- Yang, W.; Hullavarad, S.S.; Nagaraj, B.; Takeuchi, I.; Sharma, R.P.; Venkatesan, T.; Vispute, R.D.; Shen, H. Compositionally-tuned epitaxial cubic MgxZn1−xO on Si(100) for deep ultraviolet photodetectors. Appl. Phys. Lett. 2003, 82, 3424–3427. [Google Scholar] [CrossRef]
- Yang, H.; Li, Y.; Norton, D.P.; Pearton, S.J.; Jung, S.; Ren, F.; Boatner, L.A. Characteristics of unannealed ZnMgO/ZnO p-n junctions on bulk (100) ZnO substrates. Appl. Phys. Lett. 2005, 86, 172103–172106. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, Y.; Shen, D.; Lu, Y.; Zhang, J.; Fan, X.J. Structural and optical properties of MgxZn1−xO thin films prepared by the sol–gel method. J. Cryst. Growth 2002, 234, 427–430. [Google Scholar] [CrossRef]
- Wang, Y.S.; Thomas, P.J.; O’Brien, P. Optical properties of ZnO nanocrystals doped with Cd, Mg, Mn, and Fe ions. J. Phys. Chem. B 2006, 110, 21412. [Google Scholar] [CrossRef] [PubMed]
- Sasa, S.; Ozaki, M.; Koike, K.; Yano, M.; Inoue, M. High-performance ZnO/ZnMgO field-effect transistors using a hetero-metal-insulator-semiconductor structure. Appl. Phys. Lett. 2006, 89, 53502–53505. [Google Scholar] [CrossRef]
- Ohtomo, A.; Kawasaki, M.; Ohkubo, I.; Koinuma, H.; Yasuda, T.; Segawa, Y. Structure and optical properties of ZnO/Mg0.2Zn0.8O superlattices. Appl. Phys. Lett. 1999, 75, 980–983. [Google Scholar] [CrossRef]
- Choopun, S.; Vispute, R.D.; Yang, W.; Sharma, W.R.P.; Venkatesan, T.; Shen, H. Realization of band gap above 5.0 eV in metastable cubic-phase MgxZn1−xO alloy films. Appl. Phys. Lett. 2002, 80, 1529–1532. [Google Scholar] [CrossRef]
- Yin, Z.G.; Zheng, Q.D.; Chen, S.C.; Cai, D.D.; Ma, Y.L. Controllable ZnMgO electron-transporting layers for long-term stable organic solar cells with 8.06% efficiency after one-year storage. Adv. Energy Mater. 2016, 6, 1501493. [Google Scholar] [CrossRef]
- Sha, X.J.; Tian, F.B.; Li, D.; Duan, D.F.; Chu, B.H.; Liu, Y.X.; Liu, B.B.; Cui, T. Ab initio investigation of CaO-ZnO alloys under high pressure. Sci. Rep. 2015, 5, 11003. [Google Scholar] [CrossRef] [PubMed]
- Segall, M.; Lindan, P.; Probet, M.; Pickard, C.; Hasnip, P.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Mater. 2002, 14, 2717. [Google Scholar] [CrossRef]
- Vanderbilt, D. Ultrasoft pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Li, F.; Zhang, C.W.; Zhao, M.W. Magnetic and optical properties of Cu-doped ZnO nanosheet: First-principles calculations. Physica E 2013, 53, 101–105. [Google Scholar] [CrossRef]
- Zheng, F.B.; Zhang, C.W.; Wang, P.J.; Luan, H.X. First-principles prediction of the electronic and magnetic properties of nitrogen-doped ZnO nanosheets. Solid State Commun. 2012, 152, 1199–1202. [Google Scholar] [CrossRef]
- Li, Y.L.; Zhao, X.; Fang, W.L. Structural, electronic, and optical properties of Ag-doped ZnO nanowires: First principles study. J. Phys. Chem. C 2011, 115, 3552–3557. [Google Scholar] [CrossRef]
- Sun, Z.Q.; Liao, T.; Dou, Y.H.; Hwang, S.M.; Park, M.S.; Jiang, L.; Kim, J.H.; Dou, S.X. Generalized self-assembly of scalable two-dimensional transition metal oxide nanosheets. Nat. Commun. 2014, 5, 3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.P.; Chevrier, V.L.; Ceder, G. Comparison of small polaron migration and phase separation in olivine LiMnPO4 and LiFePO4 using hybrid density functional theory. Phys. Rev. B 2011, 83, 075112. [Google Scholar] [CrossRef]
- Zhang, P.H.; Capaz, R.B.; Cohen, M.L.; Louie, S.G. Theory of sodium ordering in NaxCoO2. Phys. Rev. B 2005, 71, 153102. [Google Scholar] [CrossRef]
- Zhang, Y.G.; He, H.Y.; Pan, B.C. Structural features and electronic properties of MgO nanosheets and nanobelts. J. Phys. Chem. C 2012, 116, 23130–23135. [Google Scholar] [CrossRef]
- Wu, P.; Huang, M.; Cheng, W.J.; Tang, F.L. First-principles study of B, C, N and F doped graphene-like MgO monolayer. Physica E 2016, 81, 7–13. [Google Scholar] [CrossRef]
- Chowdhury, R.; Adhikari, S.; Rees, P. Optcial properties of silicon doped ZnO. Physica B 2010, 405, 4763–4767. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Bond Length (nm) | Bond Angle (°) | ΔE (eV) | ||
|---|---|---|---|---|---|
| dZn-O | dMg-O | θO-Zn-O | θO-Mg-O | ||
| 0 at % Mg | 0.1910 | — | 120 | — | 0 |
| 6.25 at % Mg | 0.1906 | 0.1897 | 119.37 | 120 | −2.7302 |
| 12.5 at % Mg | 0.1910 | 0.1906 | 117.79 | 118.63 | −5.4635 |
| 25 at % Mg | 0.1911 | 0.1902 | 119.37 | 119.91 | −0.6607 |
| 50 at % Mg | 0.1925 | 0.1899 | 119.061 | 119.19 | −1.4450 |
| 75 at % Mg | 0.1912 | 0.1912 | — | 119.59 | −2.3109 |
| 100 at % Mg | — | 0.1881 | — | 120 | 0 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.; Sun, D.; Tian, X.; Huang, Y. First-Principles Investigation of Phase Stability, Electronic Structure and Optical Properties of MgZnO Monolayer. Materials 2016, 9, 877. https://doi.org/10.3390/ma9110877
Tan C, Sun D, Tian X, Huang Y. First-Principles Investigation of Phase Stability, Electronic Structure and Optical Properties of MgZnO Monolayer. Materials. 2016; 9(11):877. https://doi.org/10.3390/ma9110877
Chicago/Turabian StyleTan, Changlong, Dan Sun, Xiaohua Tian, and Yuewu Huang. 2016. "First-Principles Investigation of Phase Stability, Electronic Structure and Optical Properties of MgZnO Monolayer" Materials 9, no. 11: 877. https://doi.org/10.3390/ma9110877
APA StyleTan, C., Sun, D., Tian, X., & Huang, Y. (2016). First-Principles Investigation of Phase Stability, Electronic Structure and Optical Properties of MgZnO Monolayer. Materials, 9(11), 877. https://doi.org/10.3390/ma9110877