
The Influence of Conjugated Polymer Side Chain Manipulation on the Efficiency and Stability of Polymer Solar Cells

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
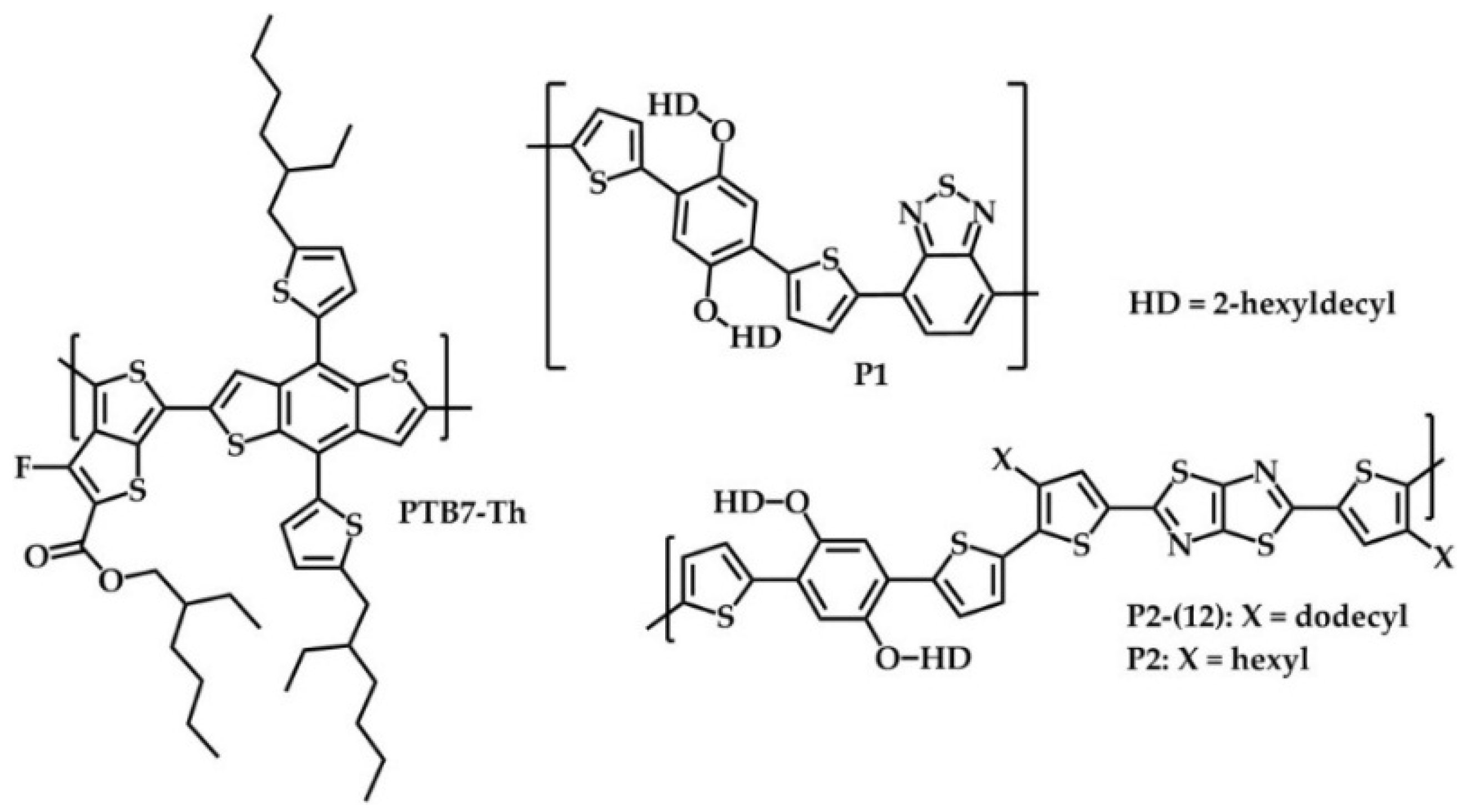
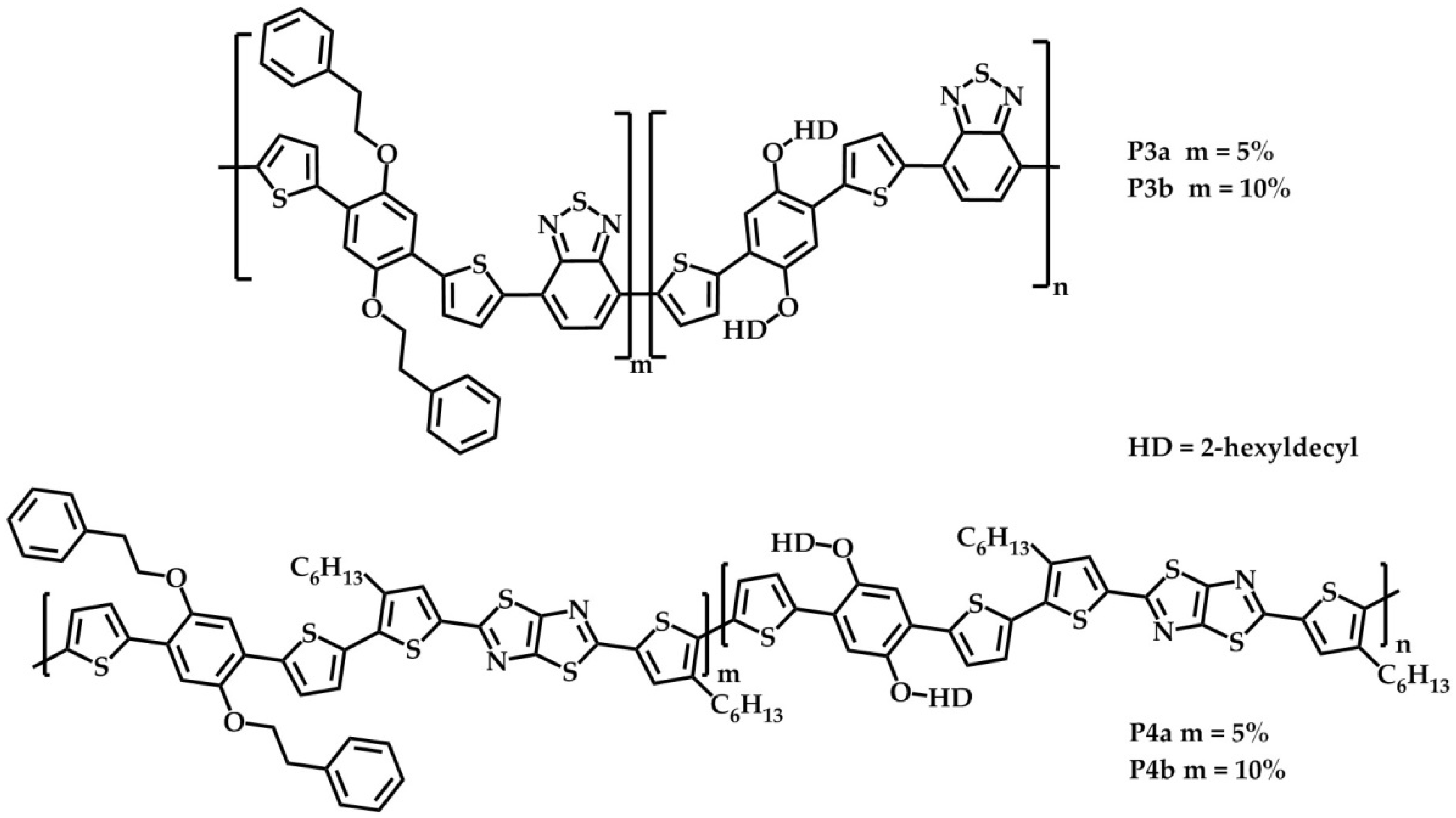
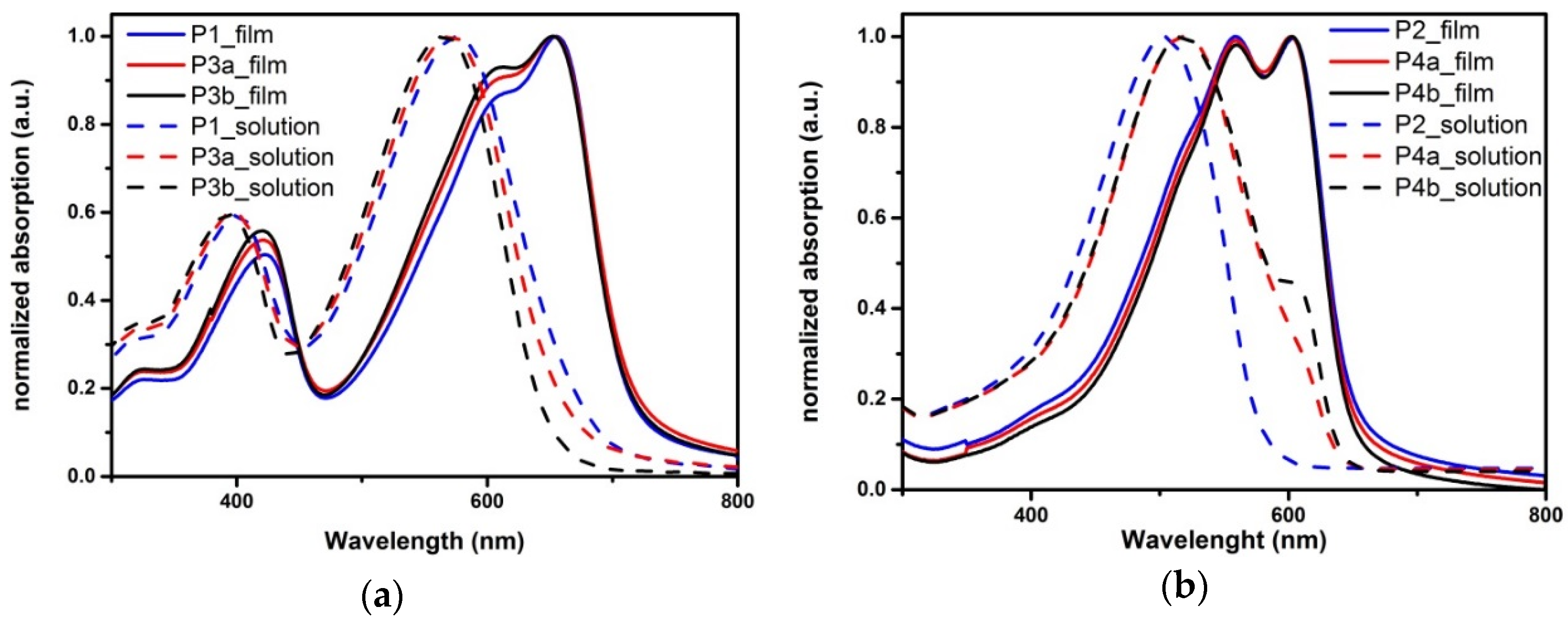
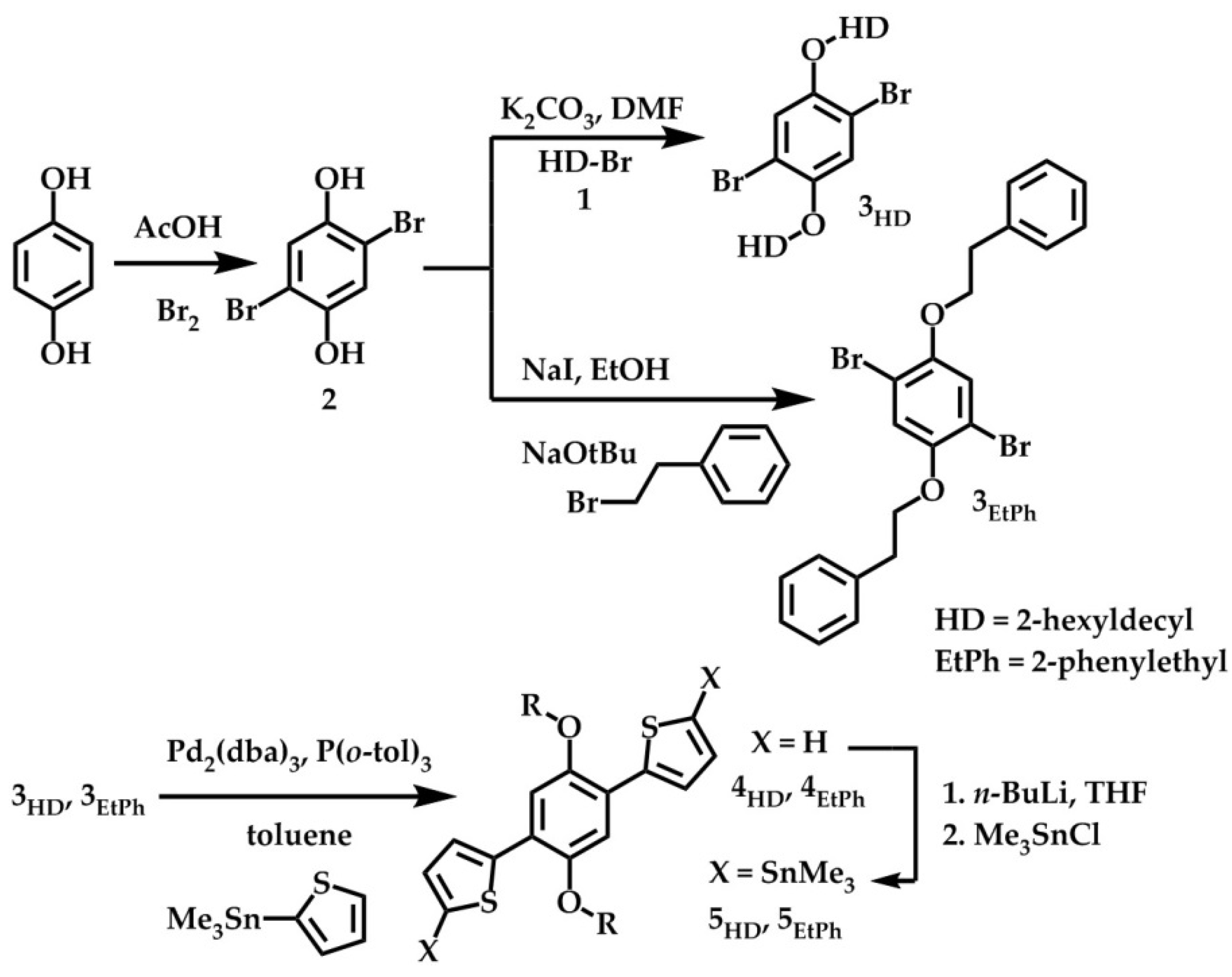
2.1. Synthesis and Material Characterization
2.2. Polymer Solar Cells
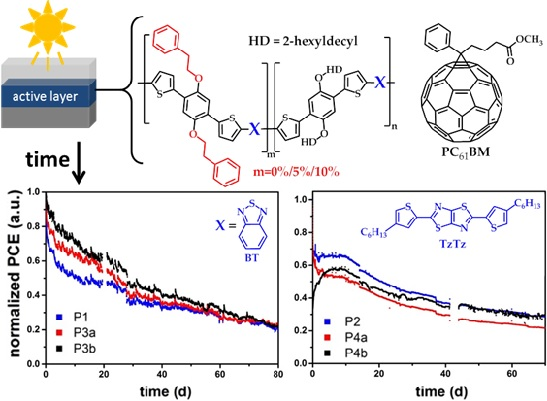
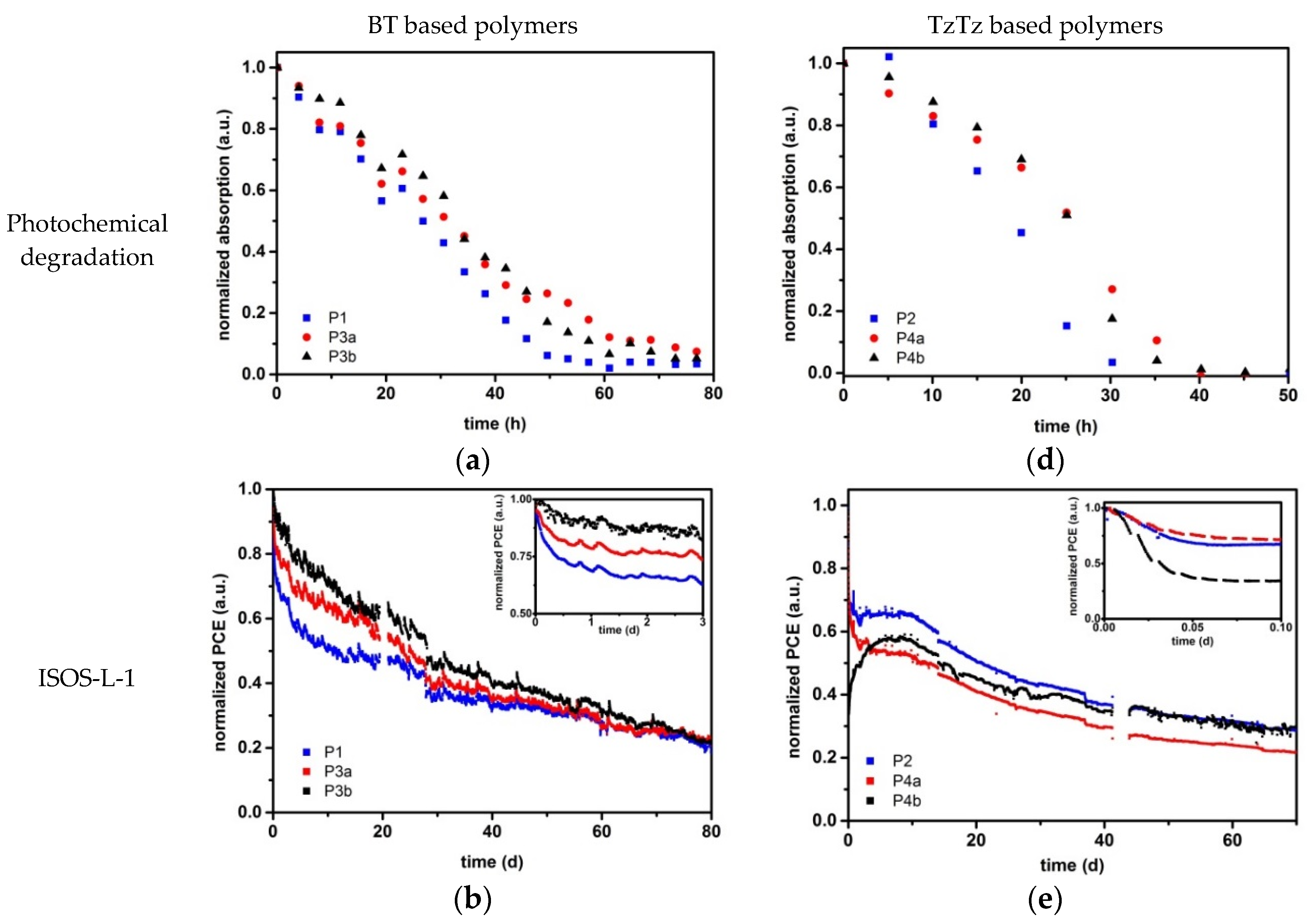
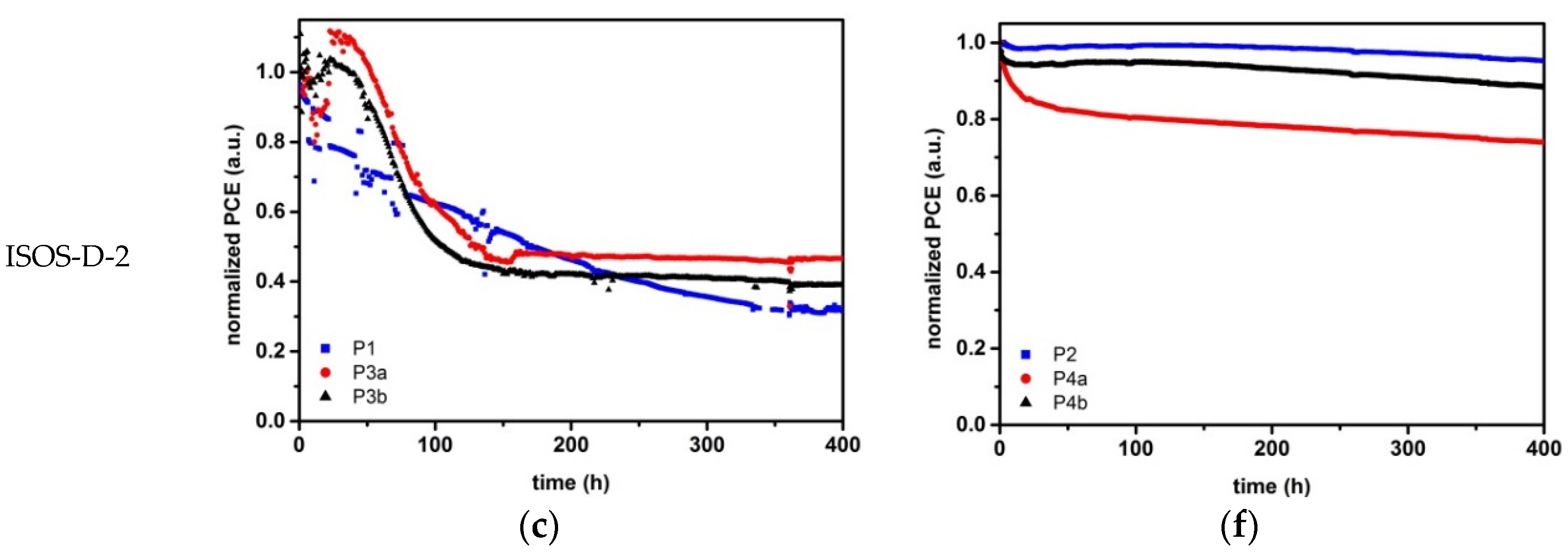
2.3. Stability Analysis
2.3.1. Material Stability
2.3.2. Device Stability
3. Experimental Section
3.1. Material and Methods
3.2. Monomer Synthesis
3.2.1. {[(2,5-Dibromo-1,4-phenylene)bis(oxy)]bis(ethane-2,1-diyl)}dibenzene (3EtPh)
3.2.2. 2,2′-(2,5-Diphenethoxy-1,4-phenylene)dithiophene (4EtPh)
3.2.3. [(2,5-Diphenethoxy-1,4-phenylene)bis(thiophene-5,2-diyl)]bis(trimethylstannane) (5EtPh)
3.3. Polymer Synthesis
3.3.1. General Procedure for the Stille Cross-Coupling Polymerization
3.3.2. Poly{[2,5-bis(2-hexyldecyloxy)phenylene]-alt-[4,7-di(thiophene-2-yl)benzo[c][1,2,5]thiadiazole]} (P1)
3.3.3. Poly{2,2′-[5,5′-(2,5-bis(2-hexyldecyloxy)-1,4-phenylene)dithiophene]-alt-[2,5-bis(4-hexyl thiophen-2-yl)thiazolo[5,4-d]thiazole]} (P2)
3.3.4. BT-Based Statistical Copolymers
3.3.5. TzTz-Based Statistical Copolymers
3.4. Device Preparation and Testing
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Günes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated polymer-based organic solar cells. Chem. Rev. 2007, 107, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Brabec, C.J.; Gowrisanker, S.; Halls, J.J.M.; Laird, D.; Jia, S.; Williams, S.P. Polymer-fullerene bulk-heterojunction solar cells. Adv. Mater. 2010, 22, 3839–3856. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-W.; Lan, S.-C.; Wei, K.-H. Organic photovoltaics. Mater. Today 2012, 15, 554–562. [Google Scholar] [CrossRef]
- Cao, W.; Xue, J. Recent progress in organic photovoltaics: Device architecture and optical design. Energy Environ. Sci. 2014, 7, 2123–2144. [Google Scholar] [CrossRef]
- Mazzio, K.A.; Luscombe, C.K. The future of organic photovoltaics. Chem. Soc. Rev. 2015, 44, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, J.; Purushothaman, B.; Chen, M.; Qin, T.; Gao, M.; Vak, D.; Scholes, F.H.; Chen, X.; Watkins, S.E.; Wilson, G.J.; et al. Organic solar cells using a high-molecular-weight benzodithiophene-benzothiadiazole copolymer with an efficiency of 9.4%. Adv. Mater. 2015, 27, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Ashraf, R.S.; Nielsen, C.B.; Collado-Fregoso, E.; Niazi, M.R.; Yousaf, S.A.; Kirkus, M.; Chen, H.-Y.; Amassian, A.; Durrant, J.R.; et al. A Thieno[3,2-b][1]benzothiophene isoindigo building block for additive- and annealing-free high-performance polymer solar cells. Adv. Mater. 2015, 27, 4702–4707. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-H.; Jhuo, H.-J.; Cheng, Y.-S.; Chen, S.-A. Fullerene derivative-doped zinc oxide nanofilm as the cathode of inverted polymer solar cells with low-bandgap polymer (PTB7-Th) for high performance. Adv. Mater. 2013, 25, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Brabec, C.J. Organic photovoltaics: Technology and market. Sol. Energy Mater. Sol. Cells 2004, 83, 273–292. [Google Scholar] [CrossRef]
- Burgues-Ceballos, I.; Stella, M.; Lacharmoise, P.; Martinez-Ferrero, E. Towards industrialization of polymer solar cells: Material processing for upscaling. J. Mater. Chem. A 2014, 2, 17711–17722. [Google Scholar] [CrossRef]
- Bundgaard, E.; Livi, F.; Hagemann, O.; Carlé, J.E.; Helgesen, M.; Heckler, I.M.; Zawacka, N.K.; Angmo, D.; Larsen-Olsen, T.T.; dos Reis Benatto, G.A.; et al. Matrix organization and merit factor evaluation as a method to address the challenge of finding a polymer material for roll coated polymer solar cells. Adv. Energy Mater. 2015, 5, 1402186. [Google Scholar] [CrossRef]
- He, Z.; Xiao, B.; Liu, F.; Wu, H.; Yang, Y.; Xiao, S.; Wang, C.; Russell, T.P.; Cao, Y. Single-junction polymer solar cells with high efficiency and photovoltage. Nat. Photonics 2015, 9, 174–179. [Google Scholar] [CrossRef]
- Carlé, J.E.; Jørgensen, M.; Krebs, F.C. Polymers for organic photovoltaics based on 1,5-bis(2-hexyldecyloxy)-naphthalene, thiophene, and benzothiadiazole. J. Photonics Energy 2011, 1. [Google Scholar] [CrossRef]
- Helgesen, M.; Carlé, J.E.; dos Reis Benatto, G.A.; Søndergaard, R.R.; Jørgensen, M.; Bundgaard, E.; Krebs, F.C. Making ends meet: Flow synthesis as the answer to reproducible high-performance conjugated polymers on the scale that roll-to-roll processing demands. Adv. Energy Mater. 2015, 5. [Google Scholar] [CrossRef]
- Livi, F.; Zawacka, N.K.; Angmo, D.; Jørgensen, M.; Krebs, F.C.; Bundgaard, E. Influence of side chain position on the electrical properties of organic solar cells based on dithienylbenzothiadiazole- alt -phenylene conjugated polymers. Macromolecules 2015, 48, 3481–3492. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, H.; You, W. Quantitatively analyzing the influence of side chains on photovoltaic properties of polymer-fullerene solar cells. J. Phys. Chem. C 2010, 114, 16793–16800. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Song, S.; Ko, S.; Choi, H.; Jeong, J.; Kim, T.; Hwang, S.; Kim, J.Y.; Woo, H.Y. Benzodithiophene-thiophene-based photovoltaic polymers with different side-chains. J. Polym. Sci. A Polym. Chem. 2015, 53, 854–862. [Google Scholar] [CrossRef]
- Cardinaletti, I.; Kesters, J.; Bertho, S.; Conings, B.; Piersimoni, F.; D’Haen, J.; Lutsen, L.; Nesladek, M.; van Mele, B.; van Assche, G.; et al. Toward bulk heterojunction polymer solar cells with thermally stable active layer morphology. J. Photonics Energy 2014, 4. [Google Scholar] [CrossRef]
- Lindqvist, C.; Bergqvist, J.; Feng, C.-C.; Gustafsson, S.; Bäcke, O.; Treat, N.D.; Bounioux, C.; Henriksson, P.; Kroon, R.; Wang, E.; et al. Fullerene nucleating agents: a route towards thermally stable photovoltaic blends. Adv. Energy Mater. 2014, 4. [Google Scholar] [CrossRef]
- Lindqvist, C.; Bergqvist, J.; Bäcke, O.; Gustafsson, S.; Wang, E.; Olsson, E.; Inganäs, O.; Andersson, M.R.; Müller, C. Fullerene mixtures enhance the thermal stability of a non-crystalline polymer solar cell blend. Appl. Phys. Lett. 2014, 104. [Google Scholar] [CrossRef]
- Rumer, J.W.; McCulloch, I. Organic photovoltaics: Crosslinking for optimal morphology and stability. Mater. Today 2015, 18, 425–435. [Google Scholar] [CrossRef]
- Gevorgyan, S.A.; Krebs, F.C. Bulk heterojunctions based on native polythiophene. Chem. Mater. 2008, 20, 4386–4390. [Google Scholar] [CrossRef]
- Bundgaard, E.; Hagemann, O.; Bjerring, M.; Nielsen, N.C.; Andreasen, J.W.; Andreasen, B.; Krebs, F.C. Removal of solubilizing side chains at low temperature: A new route to native poly(thiophene). Macromolecules 2012, 45, 3644–3646. [Google Scholar] [CrossRef]
- Verstappen, P.; Kesters, J.; D’Olieslaeger, L.; Drijkoningen, J.; Cardinaletti, I.; Vangerven, T.; Bruijnaers, B.J.; Willems, R.E.M.; D’Haen, J.; Manca, J.V.; et al. Simultaneous Enhancement of Solar Cell Efficiency and Stability by Reducing the Side Chain Density on Fluorinated PCPDTQx Copolymers. Macromolecules 2015, 48, 3873–3882. [Google Scholar] [CrossRef]
- Vandenbergh, J.; Conings, B.; Bertho, S.; Kesters, J.; Spoltore, D.; Esiner, S.; Zhao, J.; van Assche, G.; Wienk, M.M.; Maes, W.; et al. Thermal stability of poly[2-methoxy-5-(2′-phenylethoxy)-1,4-phenylenevinylene] (MPE-PPV): Fullerene bulk heterojunction solar cells. Macromolecules 2011, 44, 8470–8478. [Google Scholar] [CrossRef]
- Kesters, J.; Kudret, S.; Bertho, S.; van den Brande, N.; Defour, M.; van Mele, B.; Penxten, H.; Lutsen, L.; Manca, J.; Vanderzande, D.; et al. Enhanced intrinsic stability of the bulk heterojunction active layer blend of polymer solar cells by varying the polymer side chain pattern. Org. Electron. 2014, 15, 549–562. [Google Scholar] [CrossRef]
- Kesters, J.; Verstappen, P.; Raymakers, J.; Vanormelingen, W.; Drijkoningen, J.; D’Haen, J.; Manca, J.; Lutsen, L.; Vanderzande, D.; Maes, W. Enhanced organic solar cell stability by polymer (PCPDTBT) side chain functionalization. Chem. Mater. 2015, 27, 1332–1341. [Google Scholar] [CrossRef]
- Carlé, J.E.; Andreasen, J.W.; Jørgensen, M.; Krebs, F.C. Low band gap polymers based on 1,4-dialkoxybenzene, thiophene, bithiophene donors and the benzothiadiazole acceptor. Sol. Energy Mater. Sol. Cells 2010, 94, 774–780. [Google Scholar] [CrossRef]
- Campo, B.J.; Bevk, D.; Kesters, J.; Gilot, J.; Bolink, H.J.; Zhao, J.; Bolsée, J.-C.; Oosterbaan, W.D.; Bertho, S.; D’Haen, J.; et al. Ester-functionalized poly(3-alkylthiophene) copolymers: Synthesis, physicochemical characterization and performance in bulk heterojunction organic solar cells. Org. Electron. 2013, 14, 523–534. [Google Scholar] [CrossRef]
- Helgesen, M.; Carlé, J.E.; Krebs, F.C. Slot-die coating of a high performance copolymer in a readily scalable roll process for polymer solar cells. Adv. Energy Mater. 2013, 3, 1664–1669. [Google Scholar] [CrossRef]
- Danley, R.L.; Caulfield, P.A.; Aubuchon, S.R. A rapid-scanning differential scanning calorimeter. Am. Lab. 2008, 40, 9–11. [Google Scholar]
- Ghoos, T.; van Den Brande, N.; Defour, M.; Brassinne, J.; Fustin, C.A.; Gohy, J.F.; Hoeppener, S.; Schubert, U.S.; Vanormelingen, W.; Lutsen, L.; et al. Amphiphilic N-methylimidazole-functionalized diblock copolythiophenes. Eur. Polym. J. 2014, 53, 206–214. [Google Scholar] [CrossRef]
- Tromholt, T.; Madsen, M.V.; Carlé, J.E.; Helgesen, M.; Krebs, F.C. Photochemical stability of conjugated polymers, electron acceptors and blends for polymer solar cells resolved in terms of film thickness and absorbance. J. Mater. Chem. 2012, 22, 7592–7601. [Google Scholar] [CrossRef]
- Reese, M.O.; Gevorgyan, S.A.; Jørgensen, M.; Bundgaard, E.; Kurtz, S.R.; Ginley, D.S.; Olson, D.C.; Lloyd, M.T.; Morvillo, P.; Katz, E.A.; et al. Consensus stability testing protocols for organic photovoltaic materials and devices. Sol. Energy Mater. Sol. Cells 2011, 95, 1253–1267. [Google Scholar] [CrossRef]
- Jeong, J.; Seo, J.; Nam, S.; Han, H.; Kim, H.; Anthopoulos, T.D.; Bradley, D.D.C.; Kim, Y. Significant stability enhancement in high-efficiency Polymer:Fullerene bulk heterojunction solar cells by blocking ultraviolet photons from solar light. Adv. Sci. 2015. [Google Scholar] [CrossRef]
- Lilliedal, M.R.; Medford, A.J.; Madsen, M.V.; Norrman, K.; Krebs, F.C. The effect of post-processing treatments on inflection points in currentvoltage curves of roll-to-roll processed polymer photovoltaics. Sol. Energy Mater. Sol. Cells 2010, 94, 2018–2031. [Google Scholar] [CrossRef]
- Rivaton, A.; Chambon, S.; Manceau, M.; Gardette, J.L.; Lemaître, N.; Guillerez, S. Light-induced degradation of the active layer of polymer-based solar cells. Polym. Degrad. Stab. 2010, 95, 278–284. [Google Scholar] [CrossRef]
- Bertho, S.; Haeldermans, I.; Swinnen, A.; Moons, W.; Martens, T.; Lutsen, L.; Vanderzande, D.; Manca, J.; Senes, A.; Bonfiglio, A. Influence of thermal ageing on the stability of polymer bulk heterojunction solar cells. Sol. Energy Mater. Sol. Cells 2007, 91, 385–389. [Google Scholar] [CrossRef]
- Bard, A.; Faulkner, L. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2012. [Google Scholar]
- Trasatti, S. The absolute electrode potential: An explanatory note. Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Krebs, F.C.; Søndergaard, R.; Jørgensen, M. Printed metal back electrodes for R2R fabricated polymer solar cells studied using the LBIC technique. Sol. Energy Mater. Sol. Cells 2011, 95, 1348–1353. [Google Scholar] [CrossRef]
- Hösel, M.; Søndergaard, R.R.; Jørgensen, M.; Krebs, F.C. Fast Inline roll-to-roll printing for Indium-Tin-Oxide-free polymer solar cells using automatic registration. Energy Technol. 2013, 1, 102–107. [Google Scholar] [CrossRef]
- Larsen-Olsen, T.T.; Søndergaard, R.R.; Norrman, K.; Jørgensen, M.; Krebs, F.C. All printed transparent electrodes through an electrical switching mechanism: A convincing alternative to indium-tin-oxide, silver and vacuum. Energy Environ. Sci. 2012, 5, 9467–9471. [Google Scholar] [CrossRef] [Green Version]
- Dam, H.F.; Krebs, F.C. Simple roll coater with variable coating and temperature control for printed polymer solar cells. Sol. Energy Mater. Sol. Cells 2012, 97, 191–196. [Google Scholar] [CrossRef]
- Carlé, J.E.; Andersen, T.R.; Helgesen, M.; Bundgaard, E.; Jørgensen, M.; Krebs, F.C. A laboratory scale approach to polymer solar cells using one coating/printing machine, flexible substrates, no ITO, no vacuum and no spincoating. Sol. Energy Mater. Sol. Cells 2013, 108, 126–128. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | EtPh 1 | Mn 2 | PDI 2 | λonset 3 Solution | λonset 3 Film | ΔEop 3 | ΔEec 4 | HOMO 4 | LUMO 4 | Rd 5 |
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 0% | 42 | 1.9 | 669 | 713 | 1.73 | 2.07 | −5.37 | −3.30 | 1.66 |
| P3a | 5% | 39 | 2.1 | 656 | 715 | 1.73 | 2.03 | −5.34 | −3.31 | 1.63 |
| P3b | 10% | 43 | 2.1 | 645 | 717 | 1.73 | 1.99 | −5.31 | −3.32 | 1.69 |
| P2 | 0% | 44 | 1.9 | 580 | 650 | 1.91 | 2.24 | −5.24 | −3.01 | 2.82 |
| P4a | 5% | 30 | 3.1 | 640 | 647 | 1.91 | 2.21 | −5.20 | −2.99 | 2.60 |
| P4b | 10% | 78 | 1.5 | 640 | 646 | 1.91 | 2.22 | −5.21 | −2.99 | 2.13 |
| Polymer | Method 2 | Voc (V) | Jsc (mA/cm2) | FF | PCE (Best) (%) |
|---|---|---|---|---|---|
| P1 | RC 3 | 0.75 | 7.83 | 0.47 | 2.81 (3.05) |
| SC 4 | 0.74 | 10.38 | 0.56 | 4.31 (4.42) | |
| P3a | RC 3 | 0.74 | 6.52 | 0.52 | 2.51 (2.65) |
| SC 4 | 0.72 | 10.22 | 0.56 | 4.15 (4.54) | |
| P3b | RC 3 | 0.73 | 5.76 | 0.50 | 2.10 (2.17) |
| SC 4 | 0.71 | 9.41 | 0.55 | 3.68 (4.09) | |
| P2 | RC 5 | 0.69 | 6.69 | 0.61 | 2.67 (2.73) |
| SC 6 | 0.72 | 6.60 | 0.58 | 2.76 (2.92) | |
| P4a | RC 5 | 0.68 | 6.95 | 0.59 | 2.59 (2.72) |
| SC 6 | 0.65 | 8.57 | 0.55 | 3.06 (3.06) | |
| P4b | RC 5 | 0.69 | 7.00 | 0.60 | 2.66 (2.76) |
| SC 6 | 0.61 | 8.15 | 0.63 | 3.13 (3.21) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heckler, I.M.; Kesters, J.; Defour, M.; Madsen, M.V.; Penxten, H.; D’Haen, J.; Van Mele, B.; Maes, W.; Bundgaard, E. The Influence of Conjugated Polymer Side Chain Manipulation on the Efficiency and Stability of Polymer Solar Cells. Materials 2016, 9, 181. https://doi.org/10.3390/ma9030181
Heckler IM, Kesters J, Defour M, Madsen MV, Penxten H, D’Haen J, Van Mele B, Maes W, Bundgaard E. The Influence of Conjugated Polymer Side Chain Manipulation on the Efficiency and Stability of Polymer Solar Cells. Materials. 2016; 9(3):181. https://doi.org/10.3390/ma9030181
Chicago/Turabian StyleHeckler, Ilona M., Jurgen Kesters, Maxime Defour, Morten V. Madsen, Huguette Penxten, Jan D’Haen, Bruno Van Mele, Wouter Maes, and Eva Bundgaard. 2016. "The Influence of Conjugated Polymer Side Chain Manipulation on the Efficiency and Stability of Polymer Solar Cells" Materials 9, no. 3: 181. https://doi.org/10.3390/ma9030181
APA StyleHeckler, I. M., Kesters, J., Defour, M., Madsen, M. V., Penxten, H., D’Haen, J., Van Mele, B., Maes, W., & Bundgaard, E. (2016). The Influence of Conjugated Polymer Side Chain Manipulation on the Efficiency and Stability of Polymer Solar Cells. Materials, 9(3), 181. https://doi.org/10.3390/ma9030181