
Shape-Dependent Single-Electron Levels for Au Nanoparticles



Abstract
:1. Introduction
2. Results
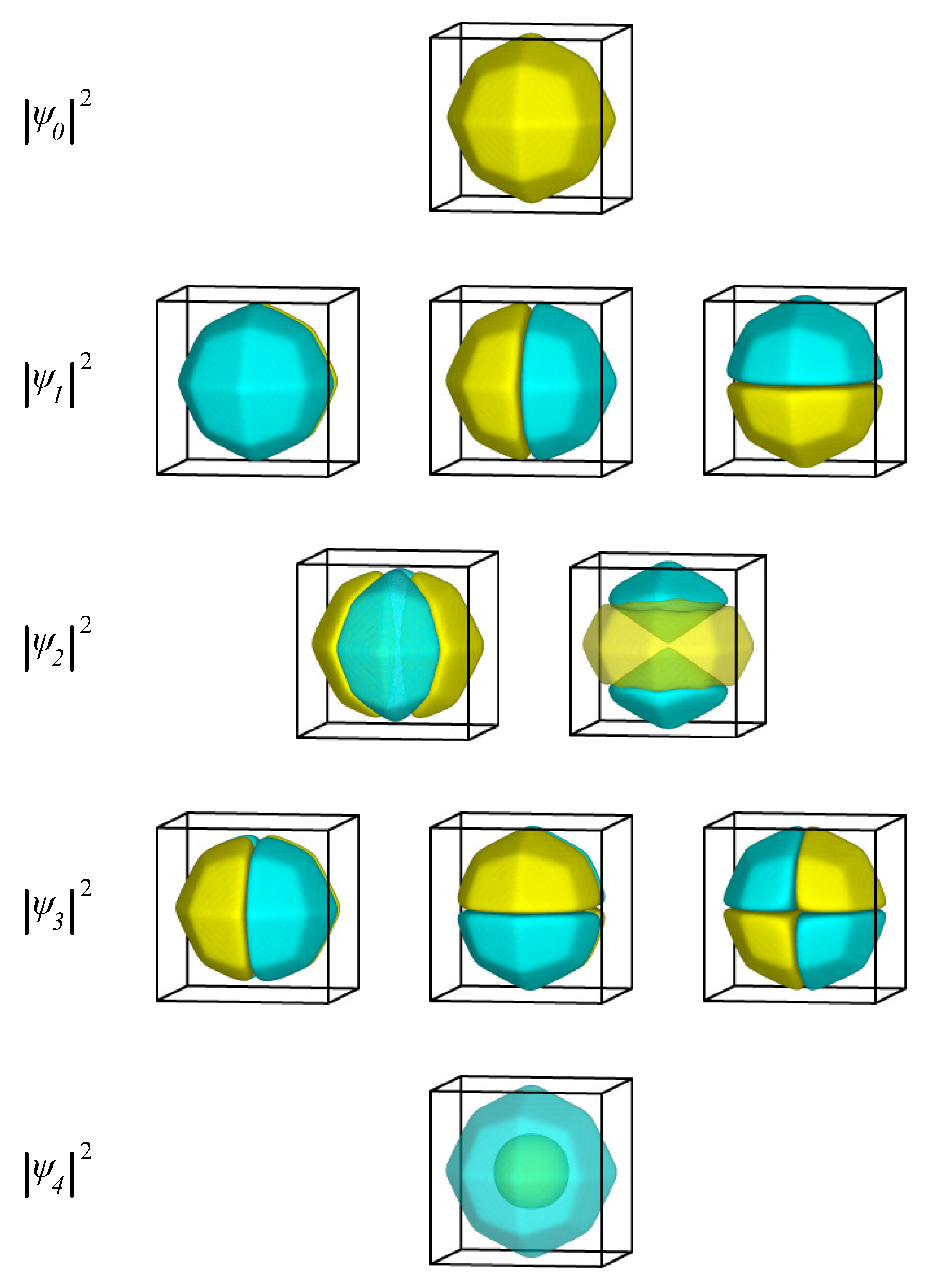
2.1. Single-Electron Levels for Typical Nanoparticle Shapes
2.1.1. Cubic Nanoparticle
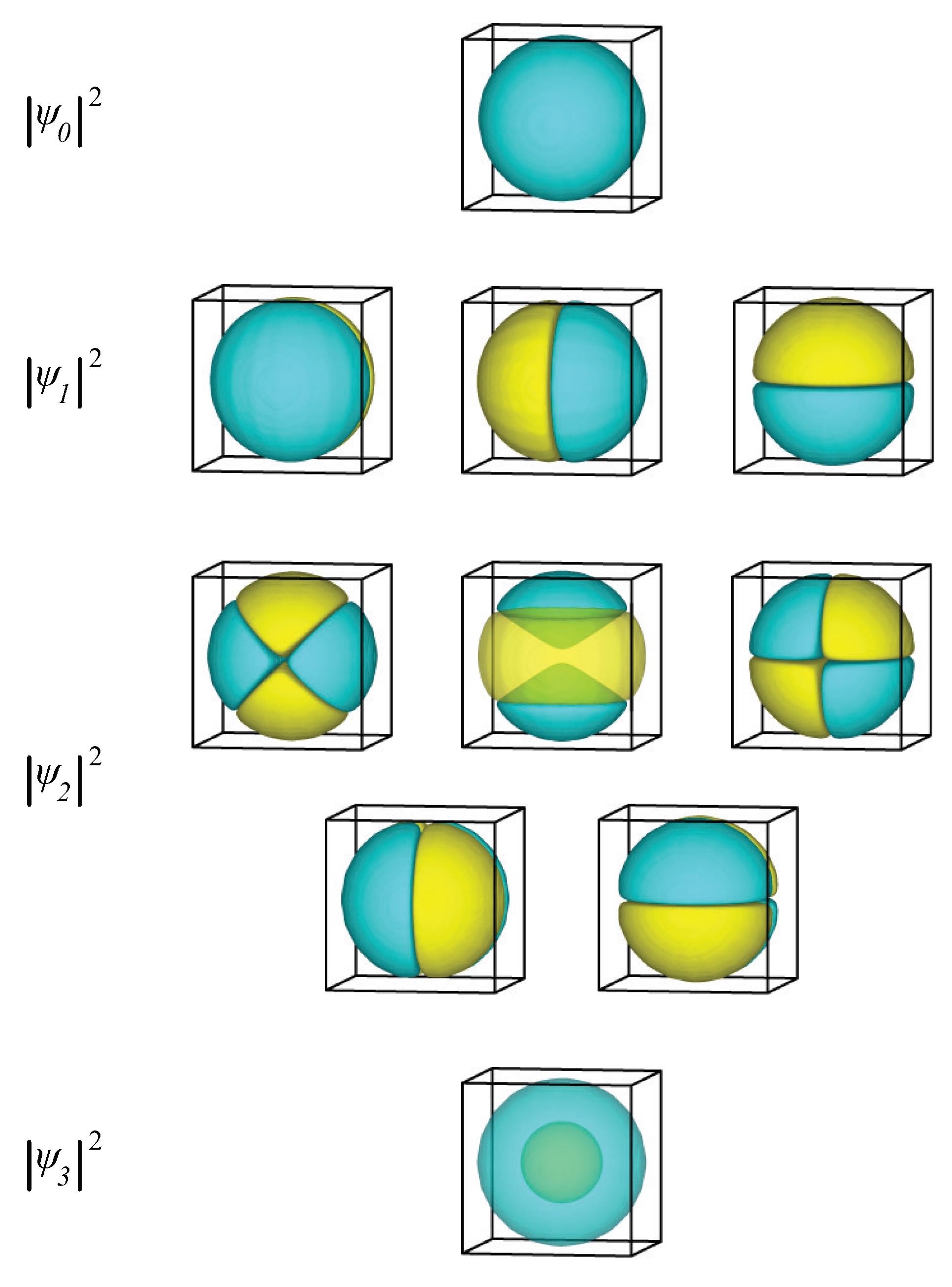
2.1.2. Spherical Nanoparticle
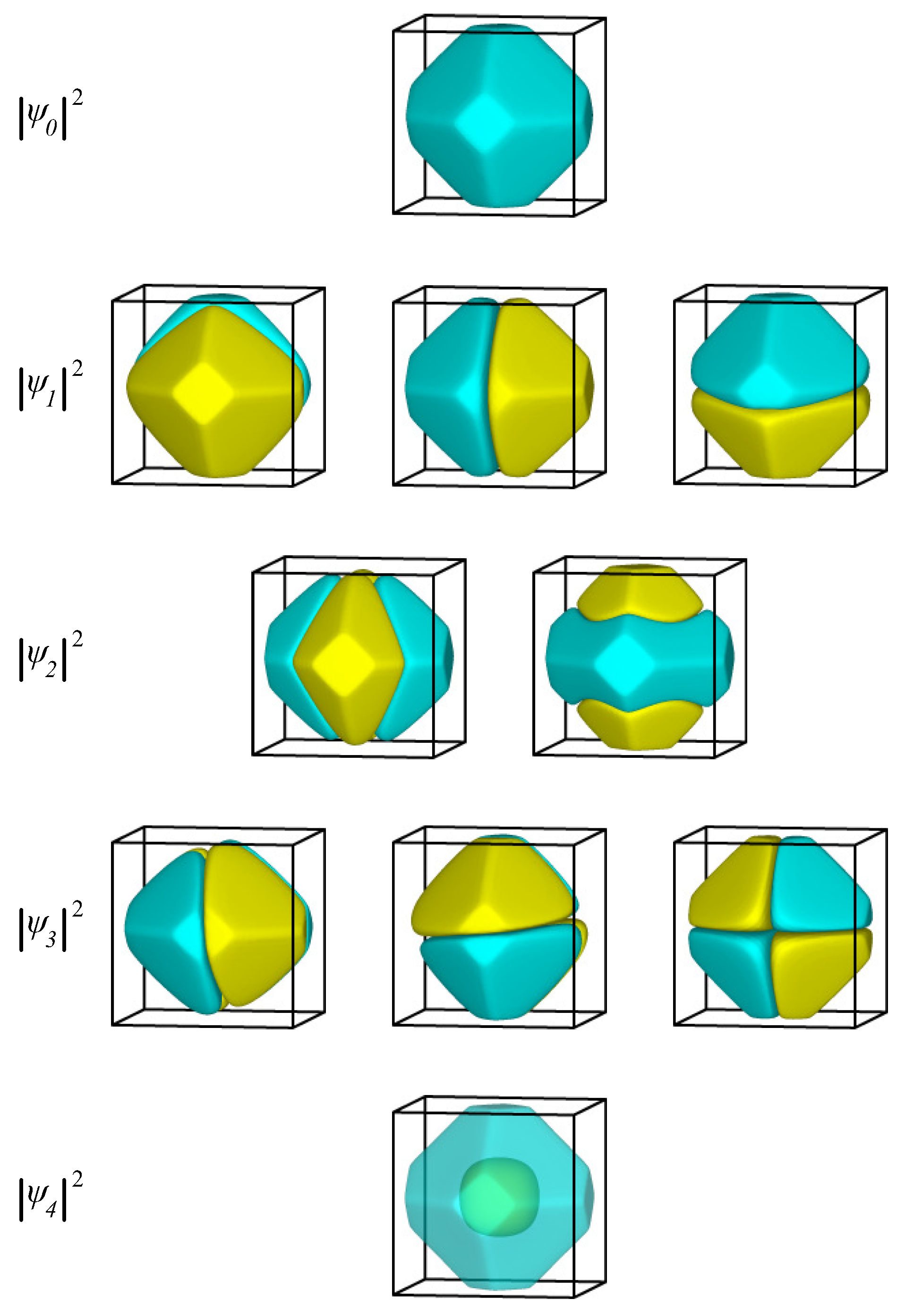
2.1.3. Single Electron in Equilibrium-Shaped Au Nanoparticles
2.1.4. Au Nanoparticles with Strong Interactions with Their Surroundings
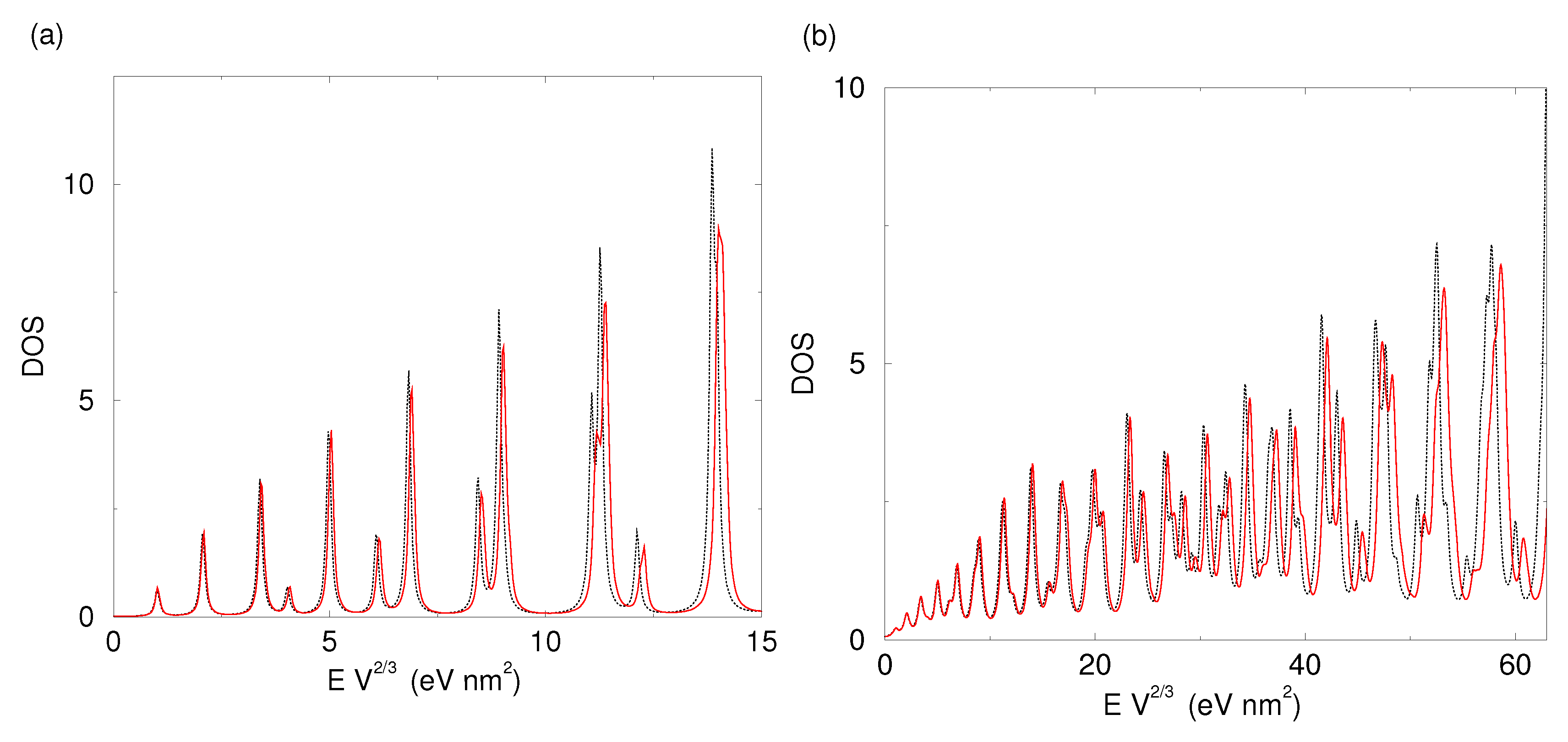
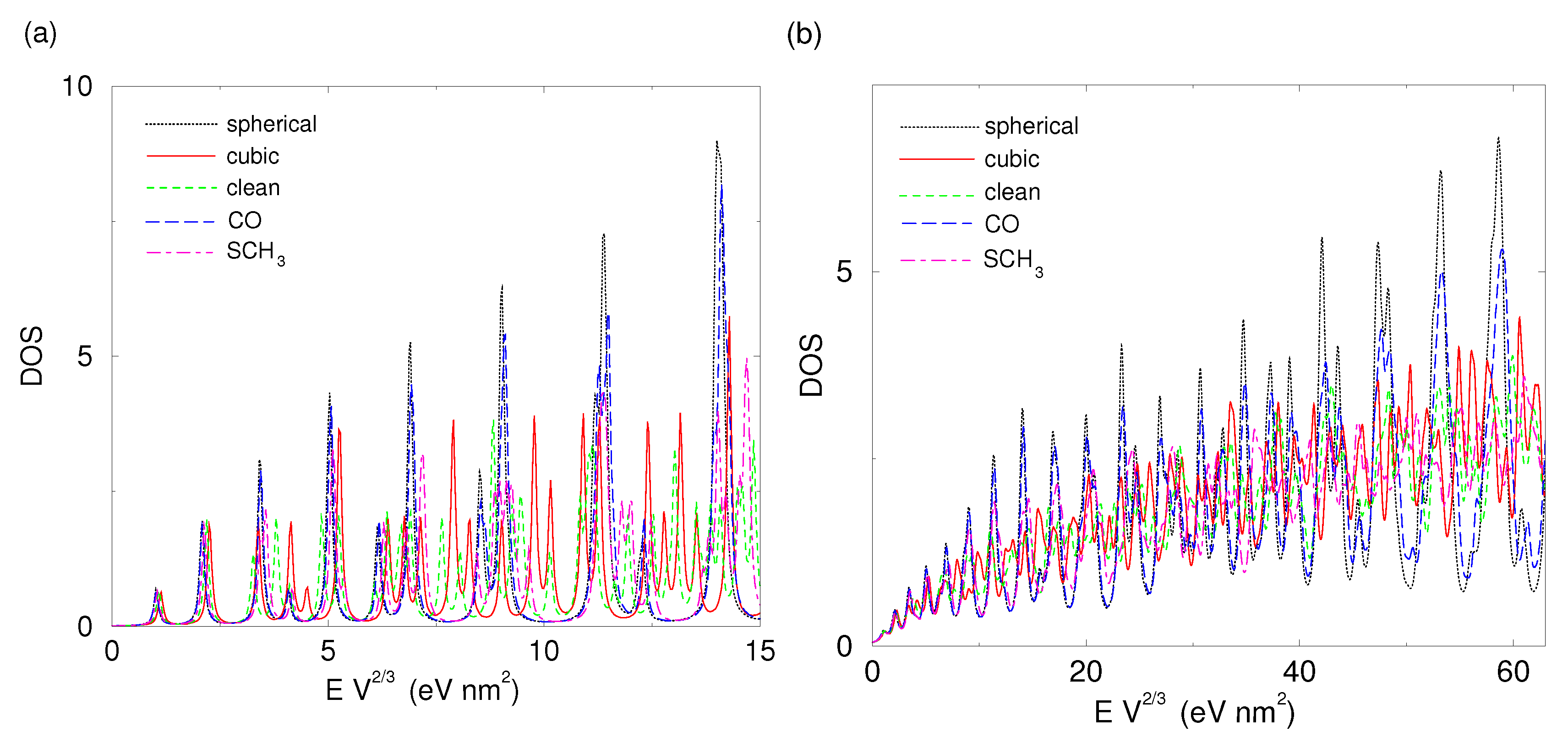
2.2. Shape-Dependent Electronic Structure
3. Materials and Methods
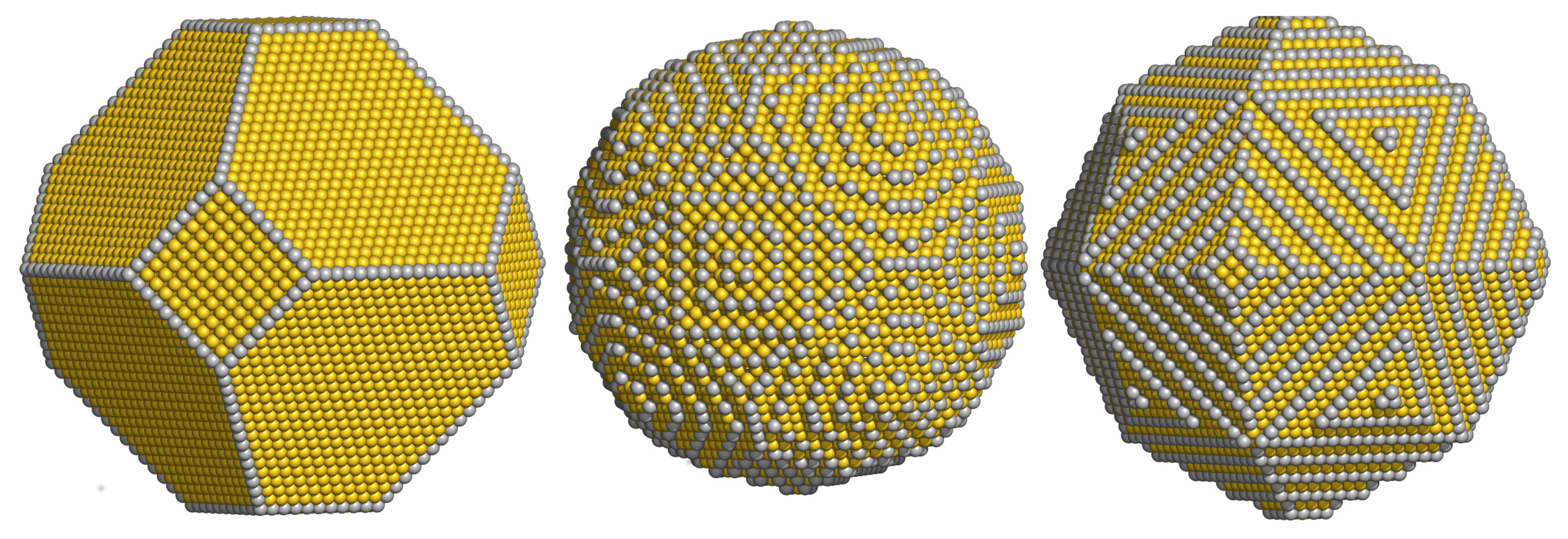
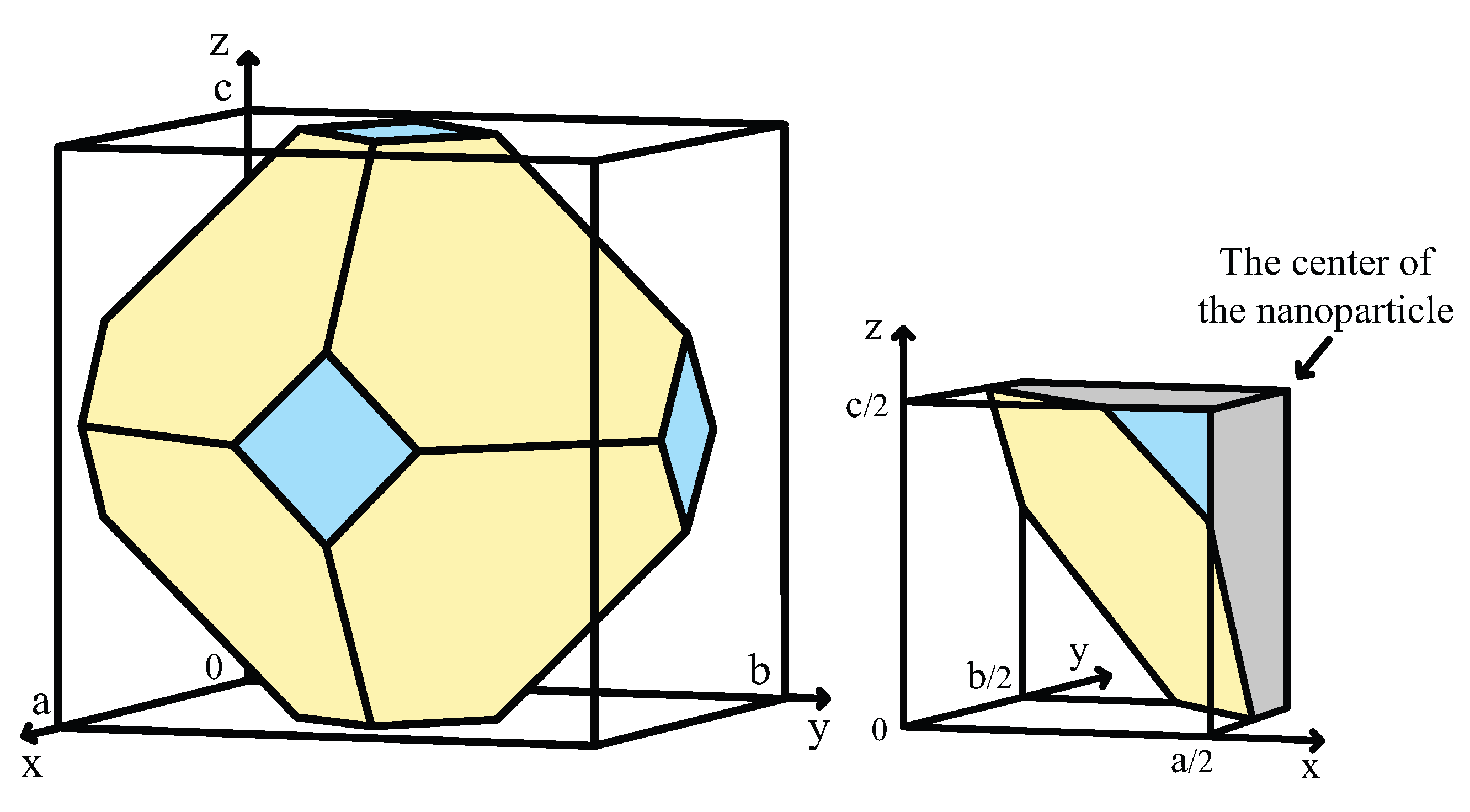
3.1. Atomistic Wulff Construction
3.2. Numerical Solution of Schrödinger’s Equation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DFT | Density-functional Theory |
| DOS | Density of states |
References
- Sun, Y.; Xia, Y. Shape-Controlled Synthesis of Gold and Silver Nanoparticles. Science 2002, 298, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Grzelczak, M.; Pérez-Juste, J.; Mulvaney, P.; Liz-Marzán, L. Shape control in gold nanoparticle synthesis. Chem. Soc. Rev. 2008, 37, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, S.; Behafarid, F.; Croy, J.R.; Ono, L.K.; Li, L.; Yang, J.C.; Frenkel, A.I.; Cuenya, B.R. Shape-Dependent Catalytic Properties of Pt Nanoparticles. J. Am. Chem. Soc. 2010, 132, 15714–15719. [Google Scholar] [CrossRef] [PubMed]
- Kovács, G.; Fodor, S.; Vulpoi, A.; Schrantz, K.; Dombi, A.; Hernádi, K.; Danciu, V.; Pap, Z.; Baia, L. Polyhedral Pt vs. spherical Pt nanoparticles on commercial titanias: Is shape tailoring a guarantee of achieving high activity? J. Catal. 2015, 325, 156–167. [Google Scholar] [CrossRef]
- Vajda, K.; Saszet, K.; Kedves, E.; Kása, Z.; Danciu, V.; Baia, L.; Magyari, K.; Hernádi, K.; Kovács, G.; Pap, Z. Shape-controlled agglomeration of TiO2 nanoparticles. New insights on polycrystallinity vs. single crystals in photocatalysis. Ceram. Int. 2016, 42, 3077–3087. [Google Scholar] [CrossRef]
- Remediakis, I.N.; Lopez, N.; Nørskov, J.K. CO Oxidation on Rutile-Supported Au Nanoparticles. Angew. Chem. Int. Ed. 2005, 44, 1824–1826. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.S.; Lin, X.M.; Curtiss, L.A. Equilibrium Morphology of Face-Centered Cubic Gold Nanoparticles >3 nm and the Shape Changes Induced by Temperature. J. Phys. Chem. B 2005, 109, 24465–24472. [Google Scholar] [CrossRef] [PubMed]
- Fasi, A.; Palinko, I.; Hernadi, K.; Kiricsi, I. Formation of Au nanorods and nanoforks over MgO support. React. Kinet. Catal. Lett. 2006, 87, 263–268. [Google Scholar] [CrossRef]
- Ahmadi, T.S.; Wang, Z.L.; Green, T.C.; Henglein, A.; Sayed, M.A.E. Shape-Controlled Synthesis of Colloidal Platinum Nanoparticles. Science 2007, 272, 1924–1926. [Google Scholar] [CrossRef]
- Ruffino, F.; Bongiorno, C.; Giannazzo, F.; Roccaforte, F.; Raineri, V.; Grimaldi, M. Effect of surrounding environment on atomic structure and equilibrium shape of growing nanocrystals: Gold in/on SiO2. Nanoscale Res. Lett. 2007, 2, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2005; Chapter 18. [Google Scholar]
- Van Aert, S.; Batenburg, K.J.; Rossell, M.D.; Erni, R.; Van Tendeloo, G. Three-dimensional atomic imaging of crystalline nanoparticles. Nature 2011, 470, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, S.; Wen, J.; Scarpelli, M.E.; Pierce, B.J.; Zuo, J.M. Equilibrium shapes and triple line energy of epitaxial gold nanocrystals supported on TiO2(110). Phys. Rev. B 2010, 82. [Google Scholar] [CrossRef]
- De Heer, W.A. The physics of simple metal clusters: Experimental aspects and simple models. Rev. Mod. Phys. 1993, 65, 611–676. [Google Scholar] [CrossRef]
- Walter, M.; Akola, J.; Lopez-Acevedo, O.; Jadzinsky, P.D.; Calero, G.; Ackerson, C.J.; Whetten, R.L.; Grönbeck, H.; Häkkinen, H. A unified view of ligand-protected gold clusters as superatom complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 9157–9162. [Google Scholar] [CrossRef] [PubMed]
- Stiehler, C.; Calaza, F.; Schneider, W.D.; Nilius, N.; Freund, H.J. Molecular Adsorption Changes the Quantum Structure of Oxide-Supported Gold Nanoparticles: Chemisorption versus Physisorption. Phys. Rev. Lett. 2015, 115. [Google Scholar] [CrossRef] [PubMed]
- Pelton, M.; Aizpurua, J.; Bryant, G. Metal-nanoparticle plasmonics. Laser Photonics Rev. 2008, 2, 136–159. [Google Scholar] [CrossRef]
- West, P.; Ishii, S.; Naik, G.; Emani, N.; Shalaev, V.; Boltasseva, A. Searching for better plasmonic materials. Laser Photonics Rev. 2010, 4, 795–808. [Google Scholar] [CrossRef]
- Mie, G. Beiträge zur Optik trüber Medien, speziell kolloidaler Metallösungen. Ann. Phys. 1908, 330, 377–445. [Google Scholar] [CrossRef]
- Maier, S.A. Plasmonics: Fundamentals and Applications, 1st ed.; Springer-Verlag US: New York, NY, USA, 2007. [Google Scholar]
- Klimov, V.I.; Mikhailovski, A.A.; Xu, S.; Malko, A.; Hollingsworth, J.A.; Leatherdale, C.A.; Eisler, H.J.; Bawendi, M.G. Optical Gain and Stimulated Emission in Nanocrystal Quantum Dots. Science 2000, 290, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Link, S.; Halas, N.J. Nano-optics from sensing to waveguiding. Nat. Photonics 2007, 1, 641–648. [Google Scholar] [CrossRef]
- Kim, S.K.; Day, R.W.; Cahoon, J.F.; Kempa, T.J.; Song, K.D.; Park, H.G.; Lieber, C.M. Tuning Light Absorption in Core/Shell Silicon Nanowire Photovoltaic Devices through Morphological Design. Nano Lett. 2012, 12, 4971–4976. [Google Scholar] [CrossRef] [PubMed]
- Beqa, L.; Singh, A.K.; Khan, S.A.; Senapati, D.; Arumugam, S.R.; Ray, P.C. Gold Nanoparticle-Based Simple Colorimetric and Ultrasensitive Dynamic Light Scattering Assay for the Selective Detection of Pb(II) from Paints, Plastics, and Water Samples. ACS Appl. Mater. Interfaces 2011, 3, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Hourahine, B.; Papoff, F. The geometrical nature of optical resonances: From a sphere to fused dimer nanoparticles. Meas. Sci. Technol. 2012, 23, 084002. [Google Scholar] [CrossRef]
- Hourahine, B.; Papoff, F. Optical control of scattering, absorption and lineshape in nanoparticles. Opt. Express 2013, 21, 20322–20333. [Google Scholar] [CrossRef] [PubMed]
- Von Delft, J.; Ralph, D. Spectroscopy of discrete energy levels in ultrasmall metallic grains. Phys. Rep. 2001, 345, 61–173. [Google Scholar] [CrossRef]
- Bolotin, K.I.; Kuemmeth, F.; Pasupathy, A.N.; Ralph, D.C. Metal-nanoparticle single-electron transistors fabricated using electromigration. Appl. Phys. Lett. 2004, 84, 3154–3156. [Google Scholar] [CrossRef]
- Kuemmeth, F.; Bolotin, K.I.; Shi, S.F.; Ralph, D.C. Measurement of Discrete Energy-Level Spectra in Individual Chemically Synthesized Gold Nanoparticles. Nano Lett. 2008, 8, 4506–4512. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Nilius, N.; Freund, H.J.; Walter, M.; Frondelius, P.; Honkala, K.; Häkkinen, H. Quantum Well States in Two-Dimensional Gold Clusters on MgO Thin Films. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef] [PubMed]
- Kac, M. Can One Hear the Shape of a Drum? Am. Math. Mon. 1966, 73, 1–23. [Google Scholar] [CrossRef]
- Hearing the Shape of a Drum. Available online: https://en.wikipedia.org/wiki/Hearing_the_shape_of_a_drum (accessed on 30 January 2016).
- Tolea, F.; Tolea, M. Hearing shapes of few electrons quantum drums: A configuration-interaction study. Physica B 2015, 458, 85–91. [Google Scholar] [CrossRef]
- Lopez, N.; Nørskov, J.; Janssens, T.; Carlsson, A.; Puig-Molina, A.; Clausen, B.; Grunwaldt, J.D. The adhesion and shape of nanosized Au particles in a Au/TiO2 catalyst. J. Catal. 2004, 225, 86–94. [Google Scholar] [CrossRef]
- Remediakis, I.N.; Lopez, N.; Nørskov, J.K. CO oxidation on gold nanoparticles: Theoretical studies. Appl. Catal. A Gen. 2005, 291, 13–21. [Google Scholar] [CrossRef]
- Quintana, M.; Ke, X.; Tendeloo, G.V.; Meneghetti, M.; Bittencourt, C.; Prato, M. Light-Induced Selective Deposition of Au Nanoparticles on Single-Wall Carbon Nanotubes. ACS Nano 2010, 4, 6105–6113. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Kawasaki, T.; Hasegawa, H.; Tanji, T.; Ichihashi, M. First observation of dynamic shape changes of a gold nanoparticle catalyst under reaction gas environment by transmission electron microscopy. Surf. Interface Anal. 2008, 40, 1725–1727. [Google Scholar] [CrossRef]
- Häkkinen, H.; Walter, M.; Grönbeck, H. Divide and Protect: Capping Gold Nanoclusters with Molecular Gold-Thiolate Rings. J. Phys. Chem. B 2006, 110, 9927–9931. [Google Scholar] [CrossRef] [PubMed]
- Barmparis, G.D.; Remediakis, I.N. Dependence on CO adsorption of the shapes of multifaceted gold nanoparticles: A density functional theory. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef]
- Bittencourt, C.; Felten, A.; Douhard, B.; Colomer, J.F.; Tendeloo, G.V.; Drube, W.; Ghijsen, J.; Pireaux, J.J. Metallic nanoparticles on plasma treated carbon nanotubes: Nano2hybrids. Surf. Sci. 2007, 601, 2800–2804. [Google Scholar] [CrossRef]
- Espinosa, E.; Ionescu, R.; Bittencourt, C.; Felten, A.; Erni, R.; Tendeloo, G.V.; Pireaux, J.J.; Llobet, E. Metal-decorated multi-wall carbon nanotubes for low temperature gas sensing. Thin Solid Films 2007, 515, 8322–8327. [Google Scholar] [CrossRef]
- Barmparis, G.D.; Honkala, K.; Remediakis, I.N. Thiolate adsorption on Au(hkl) and equilibrium shape of large thiolate-covered gold nanoparticles. J. Chem. Phys. 2013, 138. [Google Scholar] [CrossRef] [PubMed]
- Marago, O.M.; Jones, P.H.; Gucciardi, P.G.; Volpe, G.; Ferrari, A.C. Optical trapping and manipulation of nanostructures. Nat. Nano 2013, 8, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, E. Electronic states and phases of KxC60 from photoemission and X-ray absorption spectroscopy. Nature 1991, 352, 603–605. [Google Scholar]
- Liu, W.; Hu, E.; Jiang, H.; Xiang, Y.; Weng, Z.; Li, M.; Fan, Q.; Yu, X.; Altman, E.I.; Wang, H. A highly active and stable hydrogen evolution catalyst based on pyrite-structured cobalt phosphosulfide. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Zur Frage der Geschwindigkeit des Wachstums und der Auflösung der Krystallflächen. Z. Kristallogr. 1901, 34, 449–530. [Google Scholar]
- Herring, C. Some Theorems on the Free Energies of Crystal Surfaces. Phys. Rev. 1951, 82. [Google Scholar] [CrossRef]
- Rosakis, P. Continuum Surface Energy from a Lattice Model. Netw. Heterog. Media 2014, 9, 453–476. [Google Scholar] [CrossRef]
- Molina, L.M.; Hammer, B. Active Role of Oxide Support during CO Oxidation at Au/MgO. Phys. Rev. Lett. 2003, 90. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.; Zapol, P. A model for the phase stability of arbitrary nanoparticles as a function of size and shape. J. Chem. Phys. 2004, 121, 4276–4283. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.S.; Curtiss, L.A. Prediction of TiO2 Nanoparticle Phase and Shape Transitions Controlled by Surface Chemistry. Nano Lett. 2005, 5, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Hadjisavvas, G.; Remediakis, I.N.; Kelires, P.C. Insights into the Shape and Faceting of Embedded Si/α-SiO2 Nanocrystals. Phys. Rev. B 2006, 74. [Google Scholar] [CrossRef]
- Kopidakis, G.; Remediakis, I.N.; Fyta, M.G.; Kelires, P.C. Atomic and electronic structure of crystalline-amorphous carbon interfaces. Diam. Relat. Mater. 2007, 16, 1875–1881. [Google Scholar] [CrossRef]
- Mittendorfer, F.; Seriani, N.; Dubay, O.; Kresse, G. Morphology of mesoscopic Rh and Pd nanoparticles under oxidizing conditions. Phys. Rev. B 2007, 76. [Google Scholar] [CrossRef]
- Soon, A.; Wong, L.; Delley, B.; Stampfl, C. Morphology of copper nanoparticles in a nitrogen atmosphere: A first-principles investigation. Phys. Rev. B 2008, 77. [Google Scholar] [CrossRef]
- Shi, H.; Stampfl, C. Shape and surface structure of gold nanoparticles under oxidizing conditions. Phys. Rev. B 2008, 77. [Google Scholar] [CrossRef]
- Cortes-Huerto, R.; Goniakowski, J.; Noguera, C. An efficient many-body potential for the interaction of transition and noble metal nano-objects with an environment. J. Chem. Phys. 2013, 138. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Dai, B.; Johnson, J.K.; Sholl, D.S. Assessing nanoparticle size effects on metal hydride thermodynamics using the Wulff construction. Nanotechnology 2009, 20. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Duan, X.; Qian, G.; Zhou, X.; Chen, D. CO Activation Pathways of Fischer Tropsch Synthesis on χ-Fe5C2 (510): Direct versus Hydrogen-Assisted CO Dissociation. J. Phys. Chem. C 2014, 118, 10170–10176. [Google Scholar] [CrossRef]
- Lodziana, Z.; Stoica, G.; Perez-Ramirez, J. Reevaluation of the Structure and Fundamental Physical Properties of Dawsonites by DFT Studies. Inorg. Chem. 2011, 50, 2590–2598. [Google Scholar] [CrossRef] [PubMed]
- Honkala, K.; Lodziana, Z.; Remediakis, I.N.; Lopez, N. Expanding and Reducing Complexity in Materials Science Models with Relevance in Catalysis and Energy. Top. Catal. 2014, 57, 14–24. [Google Scholar] [CrossRef]
- Honkala, K.; Hellman, A.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.; Nørskov, J.K. Ammonia synthesis from first-principles calculations. Science 2005, 307, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Honkala, K.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.H.; Nørskov, J.K. Insights into ammonia synthesis from first-principles. Surf. Sci. 2006, 600, 4264–4268. [Google Scholar] [CrossRef]
- Hellman, A.; Honkala, K.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.H.; Nørskov, J.K. Ammonia synthesis and decomposition on a Ru-based catalyst modeled by first-principles. Surf. Sci. 2009, 603, 1731–1739. [Google Scholar] [CrossRef]
- Vile, G.; Baudouin, D.; Remediakis, I.N.; Coperet, C.; Lopez, N.; Perez-Ramirez, J. Silver Nanoparticles for Olefin Production: New Insights into the Mechanistic Description of Propyne Hydrogenation. ChemCatChem 2013, 5, 3750–3759. [Google Scholar] [CrossRef]
- Gómez-Graña, S.; Goris, B.; Altantzis, T.; Fernández-López, C.; Carbó-Argibay, E.; Guerrero-Martínez, A.; Almora-Barrios, N.; López, N.; Pastoriza-Santos, I.; Pérez-Juste, J.; et al. Au@Ag nanoparticles: Halides stabilize 100 facets. J. Phys. Chem. Lett. 2013, 4, 2209–2216. [Google Scholar] [CrossRef]
- Almora-Barrios, N.; Novell-Leruth, G.; Whiting, P.; Liz-Marzán, L.; López, N. Theoretical description of the role of halides, silver, and surfactants on the structure of gold nanorods. Nano Lett. 2014, 14, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Barmparis, G.D.; Lodziana, Z.; Lopez, N.; Remediakis, I.N. Nanoparticle shapes by using Wulff constructions and first-principles calculations. Beilstein J. Nanotechnol. 2015, 6, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Barmparis, G.D.; Maniadaki, A.E.; Kopidakis, G.; Remediakis, I.N. Wulff construction and molecular dynamics simulations for Au nanoparticles. J. Chem. Eng. Chem. Res. 2016. accepted. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| i | (meV) | (meV) | i | (meV) | (meV) | i | (meV) | (meV) |
|---|---|---|---|---|---|---|---|---|
| 1 | 16.577 | 15.907 | 11 | 81.605 | 78.704 | 21 | 111.900 | 107.914 |
| 2 | 33.690 | 32.543 | 12 | 81.693 | 78.704 | 22 | 112.031 | 107.914 |
| 3 | 33.953 | 32.543 | 13 | 81.791 | 78.704 | 23 | 112.122 | 107.914 |
| 4 | 33.953 | 32.543 | 14 | 81.791 | 78.704 | 24 | 112.122 | 107.914 |
| 5 | 55.534 | 53.538 | 15 | 82.017 | 78.704 | 25 | 112.259 | 107.914 |
| 6 | 55.534 | 53.538 | 16 | 82.343 | 78.704 | 26 | 112.404 | 107.914 |
| 7 | 55.652 | 53.538 | 17 | 82.343 | 78.704 | 27 | 112.404 | 107.914 |
| 8 | 55.842 | 53.538 | 18 | 99.605 | 96.189 | 28 | 112.851 | 107.914 |
| 9 | 56.070 | 53.538 | 19 | 100.370 | 96.189 | 29 | 113.054 | 107.914 |
| 10 | 66.307 | 63.630 | 20 | 100.370 | 96.189 | 30 | 138.328 | 133.323 |
| i | (eV nm) | ||||
|---|---|---|---|---|---|
| Sphere | CO | SCH | Clean | Cube | |
| 0 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 1 | 1.022 | 1.068 | 1.089 | 1.105 | 1.128 |
| 2 | 1.022 | 1.068 | 1.089 | 1.105 | 1.128 |
| 3 | 1.022 | 1.069 | 1.089 | 1.105 | 1.128 |
| 4 | 2.312 | 2.402 | 2.388 | 2.183 | 2.256 |
| 5 | 2.312 | 2.402 | 2.389 | 2.183 | 2.256 |
| 6 | 2.312 | 2.402 | 2.517 | 2.721 | 2.256 |
| 7 | 2.312 | 2.446 | 2.517 | 2.721 | 3.008 |
| 8 | 2.312 | 2.446 | 2.517 | 2.721 | 3.008 |
| 9 | 2.931 | 3.071 | 3.120 | 3.030 | 3.008 |
| 10 | 3.857 | 3.981 | 4.041 | 3.774 | 3.384 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barmparis, G.D.; Kopidakis, G.; Remediakis, I.N. Shape-Dependent Single-Electron Levels for Au Nanoparticles. Materials 2016, 9, 301. https://doi.org/10.3390/ma9040301
Barmparis GD, Kopidakis G, Remediakis IN. Shape-Dependent Single-Electron Levels for Au Nanoparticles. Materials. 2016; 9(4):301. https://doi.org/10.3390/ma9040301
Chicago/Turabian StyleBarmparis, Georgios D., Georgios Kopidakis, and Ioannis N. Remediakis. 2016. "Shape-Dependent Single-Electron Levels for Au Nanoparticles" Materials 9, no. 4: 301. https://doi.org/10.3390/ma9040301
APA StyleBarmparis, G. D., Kopidakis, G., & Remediakis, I. N. (2016). Shape-Dependent Single-Electron Levels for Au Nanoparticles. Materials, 9(4), 301. https://doi.org/10.3390/ma9040301