
Utilization of Mineral Wools as Alkali-Activated Material Precursor


Abstract
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Physical Properties of the Alkali-Activated Mineral Wools
3.2. FESEM Analysis
3.3. XRD Analysis
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix

References
- Duxson, P.; Provis, J.L.; Lukey, G.C.; van Deventer, J.S.J. The role of inorganic polymer technology in the development of “green concrete”. Cem. Concr. Res. 2007, 37, 1590–1597. [Google Scholar] [CrossRef]
- Bell, J.L.; Driemeyer, P.E.; Kriven, W.M. Formation of Ceramics from Metakaolin-Based Geopolymers. Part II: K-Based Geopolymer. J. Am. Ceram. Soc. 2009, 92, 607–615. [Google Scholar] [CrossRef]
- Pacheco-Torgal, F.; Labrincha, J.A.; Leonelli, C.; Chindaprasirt, P. The fire resistance of alkali-activated cement-based concrete binders. In Handbook of Alkali-Activated Cement, Mortars and Concretes; Woodhead Publishing Series in Civil and Structural Engineering: Cambridge, UK, 2015; Volume 2015, pp. 423–461. [Google Scholar]
- Olivier, J.G.; Janssens-Maenhout, G.; Peters, J.A.H. PBL Netherlands Environmental Assessment Agency Trends in Global CO2 Emissions 2012 Report; European Commission, Joint Research Centre, Institute for Environment and Sustainability: Luxembourg, Luxembourg, 2012. [Google Scholar]
- Papadopoulos, A.M. State of the art in thermal insulation materials and aims for future developments. Energy Build. 2005, 37, 77–86. [Google Scholar] [CrossRef]
- Väntsi, O.; Kärki, T. Mineral wool waste in Europe: A review of mineral wool waste quantity, quality, and current recycling methods. J. Mater. Cycles Waste Manag. 2013, 16, 62–72. [Google Scholar] [CrossRef]
- Ali, M.H. Fire Resistant Gypsum Board Containing Mineral Wool Fibers and Method. Patent US4557973, 7 July 1987. [Google Scholar]
- Cadotte, J. Production of Water-Laid Felted Mineral Fiber Panels Including Use of Flocculating Agent. Patent 3510394, 5 May 1970. [Google Scholar]
- Cheng, A.; Lin, W.-T.; Huang, R. Application of rock wool waste in cement-based composites. Mater. Des. 2011, 32, 636–642. [Google Scholar] [CrossRef]
- Felegi, J.; Kehrer, K. Composite Fiberboard and Process of Manufacture. Patent US5134179, 1 October 1990. [Google Scholar]
- Hagerman, R. Mineral Wool Waste Cement. Patent US4662941, 5 May 1987. [Google Scholar]
- Kizinievič, O.; Balkevičius, V.; Pranckevičienė, J.; Kizinievič, V. Investigation of the usage of centrifuging waste of mineral wool melt (CMWW), contaminated with phenol and formaldehyde, in manufacturing of ceramic products. Waste Manag. 2014, 34, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-T.; Han, T.-Y.; Huang, C.-C.; Cheng, A.; Huang, R. Using Rock Wool Wastes as Partial Replacement of Cement in Cement-Based Composites. Adv. Sci. Lett. 2012, 8, 489–494. [Google Scholar] [CrossRef]
- Long, W. Mineral Fiber-Containing Paper for the Production of Gypsum Wallboard and Wallboard Product Prepared Therewith. Patent CA1192709A1, 3 September 1984. [Google Scholar]
- Pranckevičienė, J. Impact of Mineral Wool Production Waste on Properties of Sintered Ceramics. Ph.D. Thesis, Vilnius Gediminas Technical University, Vilna, Lithuania, 11 November 2011. [Google Scholar]
- Talling, B.L.O.; Sarudis, M. Raw Material Briquette for Mineral Wool Production and Process for Its Preparation and Its Use. Patent US5472917, 5 December 1995. [Google Scholar]
- Wei-Ting Lin, A.C. Rock wool wastes as a supplementary cementitious material replacement in cement-based composites. Comput. Concr. 2013, 11. [Google Scholar] [CrossRef]
- Kinnunen, P.; Yliniemi, J.; Talling, B.L.O.; Illikainen, M. Rockwool waste in fly ash geopolymer composites. Mater. Cycles Waste Manag. 2015. under review. [Google Scholar]
- Duxson, P.; Provis, J.L.; Lukey, G.C.; Mallicoat, S.W.; Kriven, W.M.; van Deventer, J.S.J. Understanding the relationship between geopolymer composition, microstructure and mechanical properties. Colloids Surf. Physicochem. Eng. Asp. 2005, 269, 47–58. [Google Scholar] [CrossRef]
- Nugteren, H.W. Secondary Industrial Minerals from Coal Fly Ash and Aluminum Anodising Waste Solutions. Ph.D. Thesis, Technical University of Delft, Delft, The Netherlands, 10 January 2010. [Google Scholar]
- Phair, J.W.; van Deventer, J.S.J. Characterization of Fly-Ash-Based Geopolymeric Binders Activated with Sodium Aluminate. Ind. Eng. Chem. Res. 2002, 41, 4242–4251. [Google Scholar] [CrossRef]
- Bernal, S.; Provis, J.; Fernández-Jiménez, A.; Krivenko, P.; Kavalerova, E.; Palacios, M.; Shi, C. Binder Chemistry—High-Calcium Alkali-Activated Materials. In Alkali Activated Materials—State-of-the Art Report; Springer: Rotterdam, The Netherlands, 2014; Volume 2014, pp. 59–91. [Google Scholar]
- Bernal, S.A.; San Nicolas, R.; Myers, R.J.; Mejía de Gutiérrez, R.; Puertas, F.; van Deventer, J.S.J.; Provis, J.L. MgO content of slag controls phase evolution and structural changes induced by accelerated carbonation in alkali-activated binders. Cem. Concr. Res. 2014, 57, 33–43. [Google Scholar] [CrossRef]
- Walling, S.A.; Kinoshita, H.; Bernal, S.A.; Collier, N.C.; Provis, J.L. Structure and properties of binder gels formed in the system Mg(OH)2-SiO2-H2O for immobilisation of Magnox sludge. Dalton Trans. Camb. Engl. 2015, 44, 8126–8137. [Google Scholar] [CrossRef] [PubMed]
- The International Centre for Diffraction Data (ICDD). The Powder Diffraction File; The International Centre for Diffraction Data (ICDD): Philadelphia, PA, USA, 2006. [Google Scholar]
- Roylance, D. Stress-Strain Curves. Available online: http://www.saylor.org/site/wp-content/uploads/2012/09/ME1022.2.4.pdf (accessed on 14 December 2015).
- Provis, J.L.; Lukey, G.C.; van Deventer, J.S.J. Do Geopolymers Actually Contain Nanocrystalline Zeolites? A Reexamination of Existing Results. Chem. Mater. 2005, 17, 3075–3085. [Google Scholar] [CrossRef]
- Van Riessen, A.; Jamieson, E.; Kealley, C.S.; Hart, R.D.; Williams, R.P. Bayer-geopolymers: An exploration of synergy between the alumina and geopolymer industries. Cem. Concr. Compos. 2013, 41, 29–33. [Google Scholar] [CrossRef]
- Myers, R.J.; L’Hôpital, E.; Provis, J.L.; Lothenbach, B. Effect of temperature and aluminium on calcium (alumino) silicate hydrate chemistry under equilibrium conditions. Cem. Concr. Res. 2015, 68, 83–93. [Google Scholar] [CrossRef]
- Myers, R.J.; Bernal, S.A.; San Nicolas, R.; Provis, J.L. Generalized structural description of calcium-sodium aluminosilicate hydrate gels: The cross-linked substituted tobermorite model. Langmuir 2013, 29, 5294–5306. [Google Scholar] [CrossRef] [PubMed]
- Provis, J.L.; Myers, R.J.; White, C.E.; Rose, V.; van Deventer, J.S.J. X-ray microtomography shows pore structure and tortuosity in alkali-activated binders. Cem. Concr. Res. 2012, 42, 855–864. [Google Scholar] [CrossRef]
- De Jong, B.H.W.S.; Brown, G.E., Jr. Polymerization of silicate and aluminate tetrahedra in glasses, melts, and aqueous solutions—I. Electronic structure of H6Si2O7, H6AlSiO71−, and H6Al2O72−. Geochim. Cosmochim. Acta 1980, 44, 491–511. [Google Scholar] [CrossRef]
- Nazari, A.; Maghsoudpour, A.; Sanjayan, J.G. Characteristics of boroaluminosilicate geopolymers. Constr. Build. Mater. 2014, 70, 262–268. [Google Scholar] [CrossRef]
- Williams, R.P.; van Riessen, A. Development of alkali activated borosilicate inorganic polymers (AABSIP). J. Eur. Ceram. Soc. 2011, 31, 1513–1516. [Google Scholar] [CrossRef]
- Alonso, S.; Palomo, A. Alkaline activation of metakaolin and calcium hydroxide mixtures: Influence of temperature, activator concentration and solids ratio. Mater. Lett. 2001, 47, 55–62. [Google Scholar] [CrossRef]
- Rovnaník, P. Effect of curing temperature on the development of hard structure of metakaolin-based geopolymer. Constr. Build. Mater. 2010, 24, 1176–1183. [Google Scholar] [CrossRef]
- Sindhunata; van Deventer, J.S.J.; Lukey, G.C.; Xu, H. Effect of Curing Temperature and Silicate Concentration on Fly-Ash-Based Geopolymerization. Ind. Eng. Chem. Res. 2006, 45, 3559–3568. [Google Scholar] [CrossRef]
- Bakharev, T.; Sanjayan, J.G.; Cheng, Y.-B. Effect of elevated temperature curing on properties of alkali-activated slag concrete. Cem. Concr. Res. 1999, 29, 1619–1625. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Palomo, J.G.; Puertas, F. Alkali-activated slag mortars: Mechanical strength behaviour. Cem. Concr. Res. 1999, 29, 1313–1321. [Google Scholar] [CrossRef]
- Provis, J.; Fernández-Jiménez, A.; Kamseu, E.; Leonelli, C.; Palomo, A. Binder Chemistry—Low-Calcium Alkali-Activated Materials. In Alkali Activated Materials—State-of-the Art Report; Springer: Rotterdam, The Netherlands, 2014; Volume 2014, pp. 93–123. [Google Scholar]
- Nugteren, H.W.; Ogundiran, M.B.; Witkamp, G.J.; Kreutzer, M. Coal Fly Ash Activated by Waste Sodium Aluminate Solutions as an Immobilizer for Hazardous Waste. In Proceedings of the 2011 World Coal Ash WOCA Conference, Denver, CO, USA, 9–11 May 2011.
- Ogundiran, M.B.; Nugteren, H.W.; Witkamp, G.J. Immobilisation of lead smelting slag within spent aluminate—Fly ash based geopolymers. J. Hazard. Mater. 2013, 248–249, 29–36. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
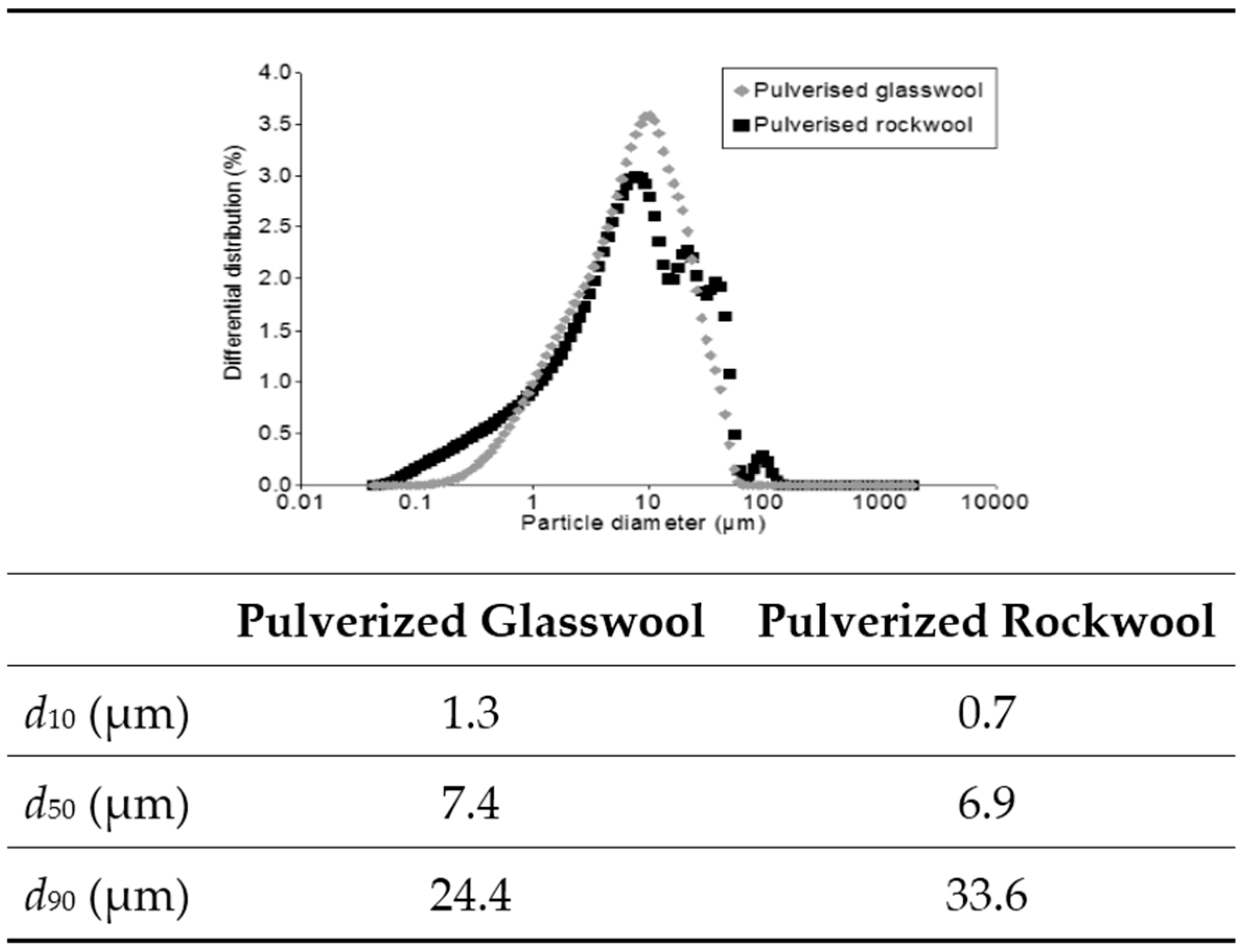{kind=link}
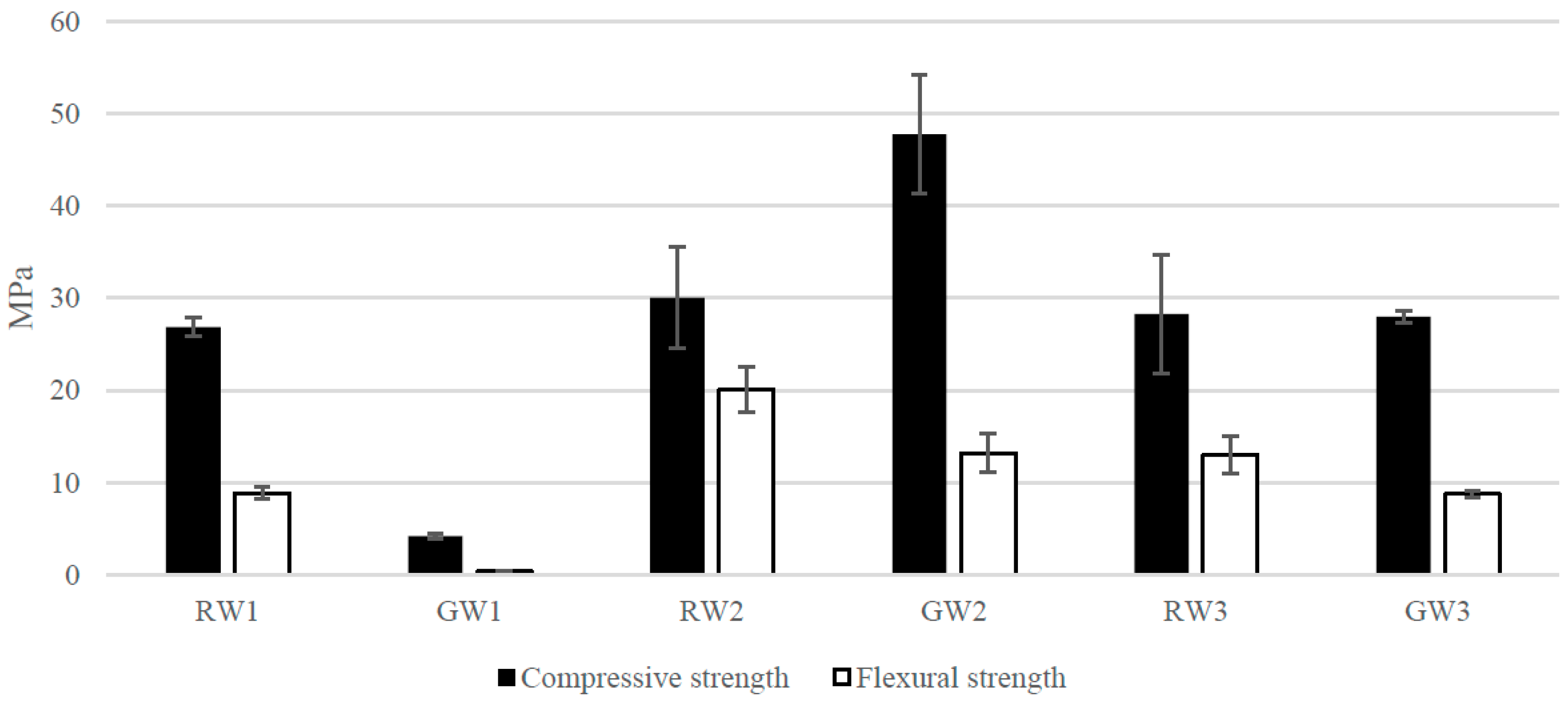{kind=link}
{kind=link}
{kind=link}
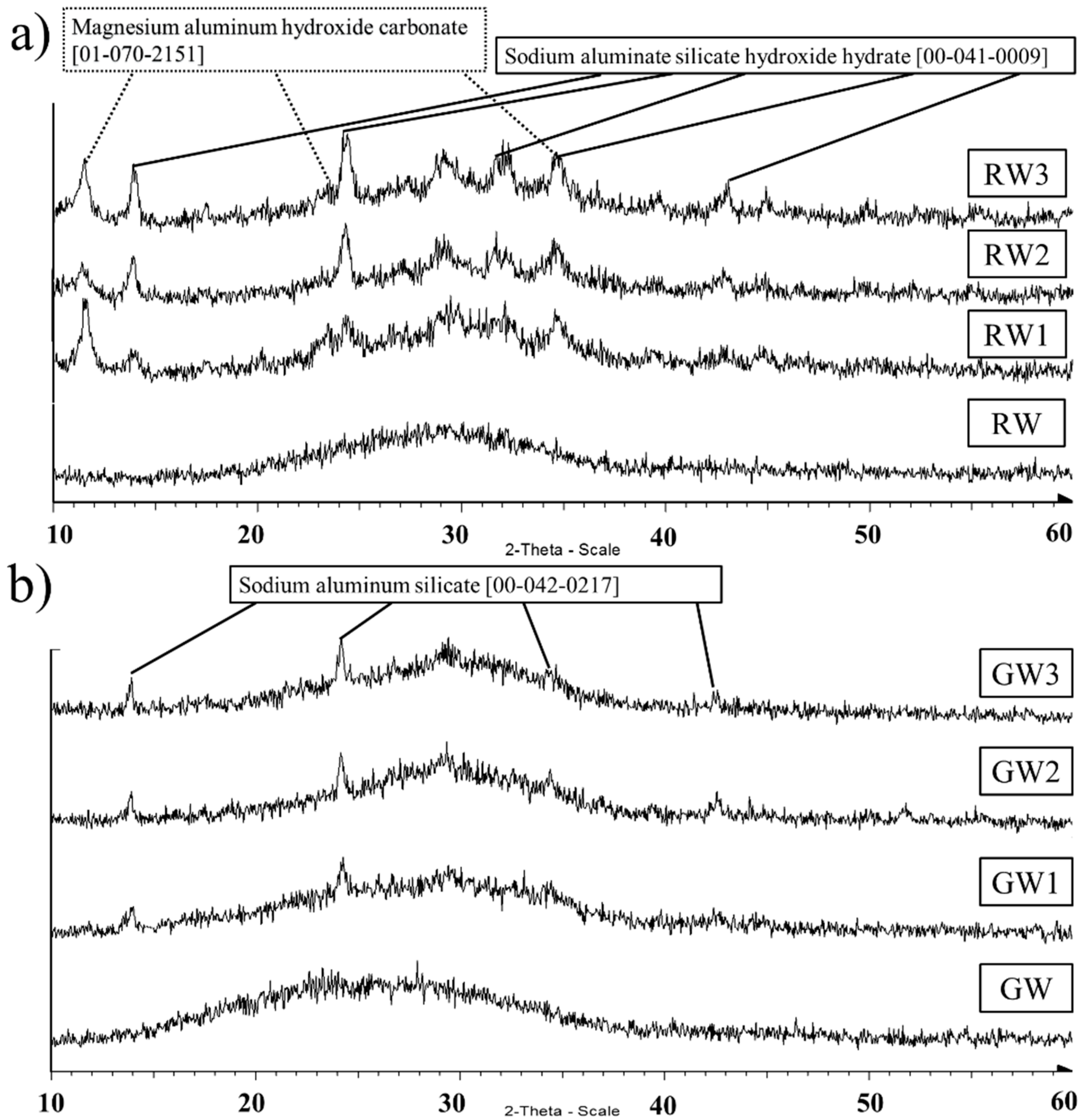{kind=link}
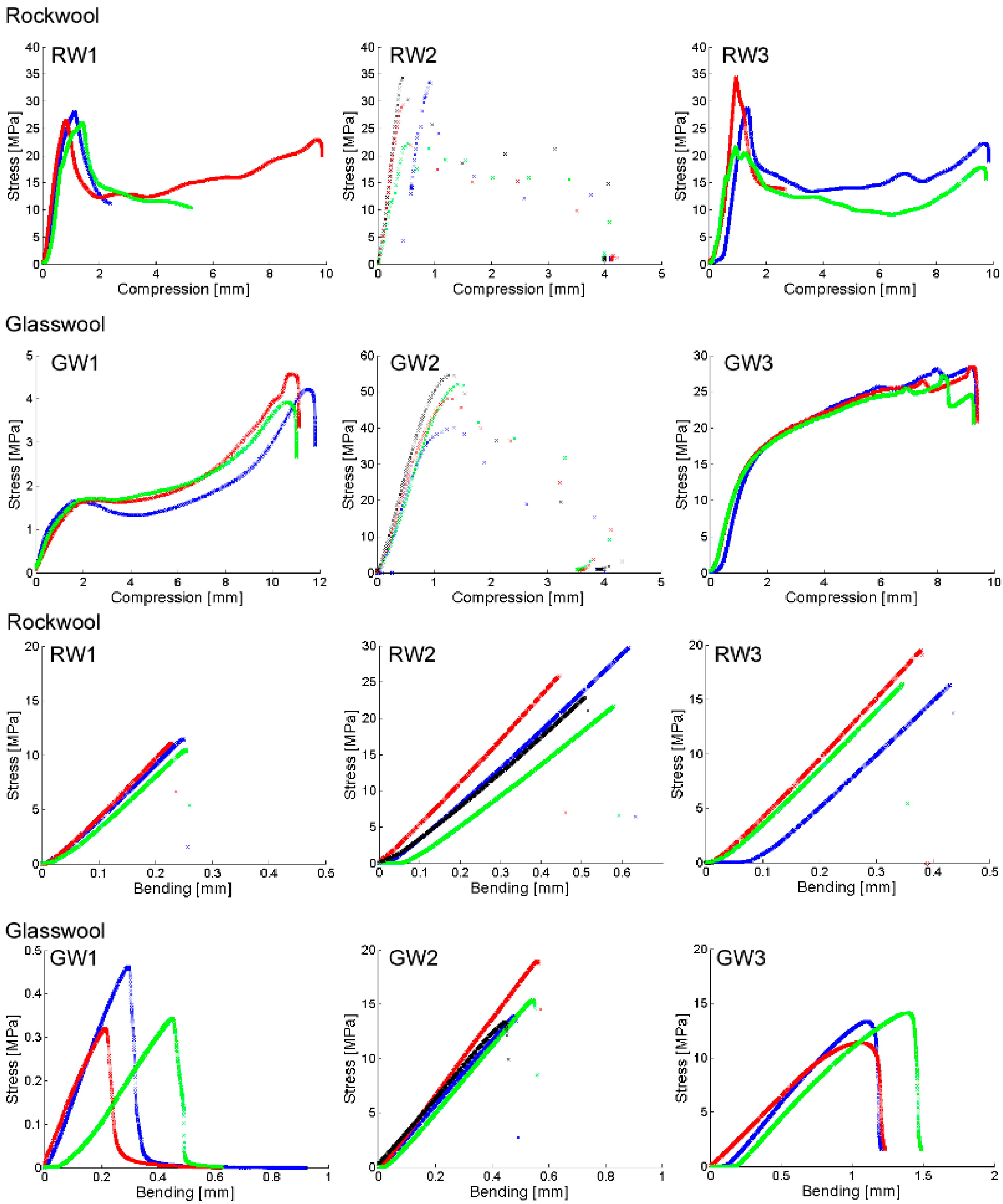{kind=link}
| Chemical Component | Rock Wool (RW) | Glass Wool (GW) |
|---|---|---|
| CaO | 17.4 | 7.1 |
| SiO2 | 40.4 | 62.4 |
| Al2O3 | 15.8 | 1.8 |
| Fe2O3 | 9.2 | 0.6 |
| Na2O | 1.4 | 16.8 |
| K2O | 0.4 | 0.9 |
| MgO | 12.6 | 2.2 |
| P2O5 | 0.1 | n.d |
| TiO2 | 0.8 | n.d |
| SO3 | n.d | 0.9 |
| Cl | n.d | 0.1 |
| Dry matter content (%) | 100.0 | 100.0 |
| Loss-on-ignition 350 °C (%) | 1.8 | 5.0 |
| Loss-on-ignition 525 °C (%) | 2.4 | 5.1 |
| Chemical Component | Rock Wool (RW) | Glass Wool (GW) |
|---|---|---|
| Ca, ICP (g/kg) | 125 | 20.1 |
| Si, ICP partial solution (g/kg) | 0.4 | 0.4 |
| Al, ICP (g/kg) | 85.6 | 0.3 |
| Fe, ICP (g/kg) | 61.3 | 0.9 |
| Na, ICP (g/kg) | 10.4 | 68.2 |
| K, ICP (g/kg) | 3.7 | 3.7 |
| Mg, ICP (g/kg) | 75.8 | 3.9 |
| P, ICP (g/kg) | 0.3 | <0.020 |
| Ti, ICP (g/kg) | 3.5 | <0.050 |
| S, ICP (g/kg) | 0.1 | 3.8 |
| Ba, ICP (g/kg) | 0.2 | 1.3 |
| Mn, ICP (g/kg) | 1 | 0.5 |
| As, ICP (mg/kg) | <3 | <3 |
| Cd, ICP (mg/kg) | <0.3 | <0.3 |
| Cr, ICP (mg/kg) | 280 | 2.3 |
| Cu, ICP (mg/kg) | 34 | 8.8 |
| Hg, CVAAS (mg/kg) | <0.04 | <0.04 |
| Ni, ICP (mg/kg) | 49 | 1.8 |
| Pb, ICP (mg/kg) | <3 | 3.7 |
| Zn, ICP (mg/kg) | 47 | 430 |
| B, ICP (mg/kg) | 8.9 | 6260 |
| Be, ICP (mg/kg) | <1 | <1 |
| Co, ICP (mg/kg) | 21 | 2 |
| Mo, ICP (mg/kg) | <1 | <1 |
| Sb, ICP (mg/kg) | <3 | <3 |
| Se, ICP (mg/kg) | <3 | <3 |
| Sn, ICP (mg/kg) | <3 | <3 |
| V, ICP (mg/kg) | 170 | <2 |
| Sample Name | RW1 | GW1 | RW2 | GW2 | RW3 | GW3 |
|---|---|---|---|---|---|---|
| Mineral wool type | Pulverized rock wool | Pulverized glass wool | Pulverized rock wool | Pulverized glass wool | Pulverized and resin removed rock wool | Pulverized and resin removed glass wool |
| Na2O (mol) | 0.7 | 7.6 | 0.7 | 7.6 | 0.7 | 7.5 |
| Al2O3 (mol) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| SiO2 (mol) | 3.6 | 20.5 | 3.6 | 21.2 | 3.6 | 21.1 |
| H2O (mol) | 8.4 | 29.5 | 8.4 | 30.3 | 8.5 | 30.4 |
| CaO (mol) | 1.7 | 2.5 | 1.7 | 2.6 | 1.7 | 2.6 |
| MgO (mol) | 1.7 | 1.1 | 1.7 | 1.1 | 1.7 | 1.1 |
| Curing (temp. and time) | 28 days in 22 °C | 28 days in 22 °C | 4 days in 50 °C and then 24 days in 22 °C | 4 days in 50 °C and then 24 days in 22 °C | 4 days in 50 °C and then 24 days in 22 °C | 4 days in 50 °C and then 24 days in 22 °C |
| Sample Code | Density (kg/m3) |
|---|---|
| RW1 | 2093 |
| GW1 | 1779 |
| RW2 | 2003 |
| GW2 | 1802 |
| RW3 | 1956 |
| GW3 | 2037 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yliniemi, J.; Kinnunen, P.; Karinkanta, P.; Illikainen, M. Utilization of Mineral Wools as Alkali-Activated Material Precursor. Materials 2016, 9, 312. https://doi.org/10.3390/ma9050312
Yliniemi J, Kinnunen P, Karinkanta P, Illikainen M. Utilization of Mineral Wools as Alkali-Activated Material Precursor. Materials. 2016; 9(5):312. https://doi.org/10.3390/ma9050312
Chicago/Turabian StyleYliniemi, Juho, Paivo Kinnunen, Pasi Karinkanta, and Mirja Illikainen. 2016. "Utilization of Mineral Wools as Alkali-Activated Material Precursor" Materials 9, no. 5: 312. https://doi.org/10.3390/ma9050312
APA StyleYliniemi, J., Kinnunen, P., Karinkanta, P., & Illikainen, M. (2016). Utilization of Mineral Wools as Alkali-Activated Material Precursor. Materials, 9(5), 312. https://doi.org/10.3390/ma9050312