
The Mechanical and Electronic Properties of Carbon-Rich Silicon Carbide



Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Round, H.J. A note on carborundum. Electr. World 1907, 49, 309. [Google Scholar]
- Zheludev, N. The life and times of the LED—A 100-year history. Nat. Photonics 2007, 1, 189–192. [Google Scholar] [CrossRef]
- Katoh, Y.; Snead, L.L.; Henager, C.H., Jr.; Hasegawa, A.; Kohyama, A.; Riccardi, B.; Hegeman, H. Current status and critical issues for development of SiC composites for fusion applications. J. Nucl. Mater. 2007, 659–671. [Google Scholar] [CrossRef]
- Harris, G.L. Properties of Silicon Carbide; INSPEC: London, UK; Institution of Electrical Engineers: London, UK, 1995. [Google Scholar]
- Hinoki, T.; Katoh, Y.; Snead, L.; Jung, H.C.; Ozawa, K.; Katsui, H.; Zhong, Z.H.; Kondo, S.; Park, Y.H.; Shih, C.H.; et al. Silicon carbide and silicon carbide composites for fusion reactor application. Mater. Trans. 2013, 54, 472–476. [Google Scholar] [CrossRef]
- Kamitani, K.; Grimsditch, M.; Nipko, J.C.; Loong, C.K.; Okada, M.; Kimura, I. The elastic constants of silicon carbide: A Brillouin-scattering study of 4H and 6H SiC single crystals. J. Appl. Phys. 1997, 82, 3152. [Google Scholar] [CrossRef]
- Sarasamak, K.; Limpijumnong, S.; Lambrecht, W.R.L. Pressure-dependent elastic constants and sound velocities of wurtzite SiC, GaN, InN, ZnO, and CdSe, and their relation to the high-pressure phase transition: A first-principles study. Phys. Rev. B 2010, 82, 035201. [Google Scholar] [CrossRef]
- Feldman, D.W.; Parker, J.H.; Choyke, W.J.; Patrick, L. Phonon Dispersion Curves by Raman Scattering in SiC, Polytypes 3C, 4H, 6H, 15R, and 21R. Phys. Rev. 1968, 173, 787. [Google Scholar] [CrossRef]
- Choyke, W.J.; Palik, E.D. Handbook of Optical Constants; Academic Press: New York, NY, USA, 1985; p. 587. [Google Scholar]
- Pizzagalli, L. Stability and mobility of screw dislocations in 4H, 2H and 3C silicon carbide. Acta. Mater. 2014, 78, 236. [Google Scholar] [CrossRef]
- Ninomiya, S.; Adachi, S. Optical Constants of 6H–SiC Single Crystals. Jpn. J. Appl. Phys. 1994, 33, 2479. [Google Scholar] [CrossRef]
- Yan, X.; Xin, Z.H.; Tian, L.J.; Yu, M. Structural and electronic properties of SimCn graphyne-like monolayers. Comput. Materi. Sci. 2015, 107, 8–14. [Google Scholar] [CrossRef]
- Zhang, X.D.; Ying, C.H.; Quan, S.Y.; Shi, G.M.; Li, Z.J. A first principles investigation on the structural, phonon, elastic and thermodynamic properties of the Si0.5Sn0.5 cubic alloy. Solid State Commun. 2012, 152, 955–959. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Xiang, G.; Gu, G.X.; Li, R.; He, D.W.; Zhang, X. Nonlinear Concentration-Dependent Electronic and Optical Properties of Si1−xGex Alloy Nanowires. J. Phys. Chem. C 2012, 116, 17934. [Google Scholar] [CrossRef]
- Zhang, Q.; Wei, Q.; Yan, H.Y.; Fan, Q.Y.; Zhu, X.M.; Zhang, J.Q.; Zhang, D.Y. Mechanical and electronic properites of P42/mnm silicon carbides. Z. Naturforschung A 2015. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T.; Qiao, L.P.; Zhao, Y.B.; Zhou, P.K.; Xing, M.J.; Zhang, J.Q.; Yao, R.H. Mechanical and electronic properties of Ca1-xMgxO alloys. Mater. Sci. Semicond. Proc. 2015, 40, 676–684. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T.; Yang, Q.; Chen, P.Y.; Xing, M.J.; Zhang, J.Q.; Yao, R.H. Prediction of novel phase of silicon and Si–Ge alloys. J. Solid State Chem. 2016, 233, 471–483. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Q.; Zhou, P.K.; Xing, M.J.; Yang, Y.T. Mechanical and electronic properties of Si, Ge and their alloys in P42/mnm structure. Mater. Sci. Semicond. Proc. 2016, 43, 187–195. [Google Scholar] [CrossRef]
- Freitas, R.R.Q.; de Brito Mota, F.; Rivelino, R.; de Castilho, C.M.C.; Kakanakova-Georgieva, A.; Gueorguiev, G.K. Spin-orbit-induced gap modification in buckled honeycomb XBi and XBi3 (X = B, Al, Ga, and In) sheets. J. Phys. Condens. Matter 2015, 27, 485306. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wang, Q.Q.; Xu, B.; Yu, D.L.; Liu, Z.Y.; Tian, Y.J.; He, J.L. Prediction of Novel SiCN compounds: First-principles calculations. J. Phys. Chem. C 2013, 117, 21943. [Google Scholar] [CrossRef]
- Zhang, X.X.; Wang, Y.C.; Lv, J.; Zhu, C.Y.; Li, Q.; Zhang, M.; Li, Q.; Ma, Y.M. First-principles structural design of superhard materials. J. Chem. Phys. 2013, 138, 114101. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892R. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the Quasi-Newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Wu, Z.H.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Wei, Q.; Yan, H.Y.; Zhang, M.G.; Zhang, D.Y.; Zhang, J.Q. A New Potential Superhard Phase of OsN2. Acta Phys. Pol. A 2014, 126, 740–746. [Google Scholar] [CrossRef]
- Karch, K.; Pavone, P.; Windl, W.; Schutt, O.; Strauch, D. Ab initio calculation of structural and lattice-dynamical properties of silicon carbide. Phys. Rev. B 1994, 50, 17054. [Google Scholar] [CrossRef]
- Madelung, O.; Rossler, U.; Schulz, M. Group IV Elements, IVIV and III-V Compounds. Part B; Springer: Berlin, Germamy, 2002; pp. 1–11. [Google Scholar]
- Lee, D.H.; Joannopoulos, J.D. Simple Scheme for Deriving Atomic Force Constants: Application to SiC. Phys. Rev. Lett. 1982, 48, 1846. [Google Scholar] [CrossRef]
- Madelung, O. Physics of Group IV and III–V Compounds, Group III, Part. A; Springer: Berlin, Germany, 1982. [Google Scholar]
- Carnahan, R.D. Elastic Properties of Silicon Carbide. J. Am. Ceram. Soc. 1968, 51, 223–224. [Google Scholar] [CrossRef]
- Lambrecht, W.R.L.; Segall, B.; Methfessel, M.; van Schilfgaarde, M. Calculated elastic constants and deformation potentials of cubic SiC. Phys. Rev. B 1991, 44, 3685. [Google Scholar] [CrossRef]
- Voigt, W. Lehrburch der Kristallphysik; Teubner: Leipzig, Germany, 1928. [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. J. Appl. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Phys. Soc. Lond. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Ding, Y.C.; Chen, M.; Gao, X.Y.; Jiang, M.H. Theoretical investigation on the electronic structure, elastic properties, and intrinsic hardness of Si2N2O. Chin. Phys. B 2012, 21, 067101. [Google Scholar] [CrossRef]
- Watt, J.P.; Peselnick, L. Clarification of the Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with hexagonal, trigonal, and tetragonal symmetries. J. Appl. Phys. 1980, 51, 1525. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the properties of polycrystalline pure plastic metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Korozlu, N.; Colakoglu, K.; Deligoz, E.; Aydin, S. The elastic and mechanical properties of MB12 (M = Zr, Hf, Y, Lu) as a function of pressure. J. Alloys Compd. 2013, 546, 157–164. [Google Scholar] [CrossRef]
- Özişik, H.; Çiftci, Y.; Çolakoglu, K.; Deligöz, E. The structural, elastic and vibrational properties of the DyX (X = P, As) compounds. Phys. Scr. 2011, 83, 035601. [Google Scholar] [CrossRef]
- Lyakhov, A.O.; Oganov, A.R. Evolutionary search for superhard materials: Methodology and applications to forms of carbon and TiO2. Phys. Rev. B 2011, 84, 092103. [Google Scholar] [CrossRef]
- Brookes, C.A.; Brookes, E.J. Diamond in perspective: A review of mechanical properties of natural diamond. Diamond Relat. Mater. 1991, 1, 13–17. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comput. Mater. Sci. 2014, 83, 27–34. [Google Scholar] [CrossRef]








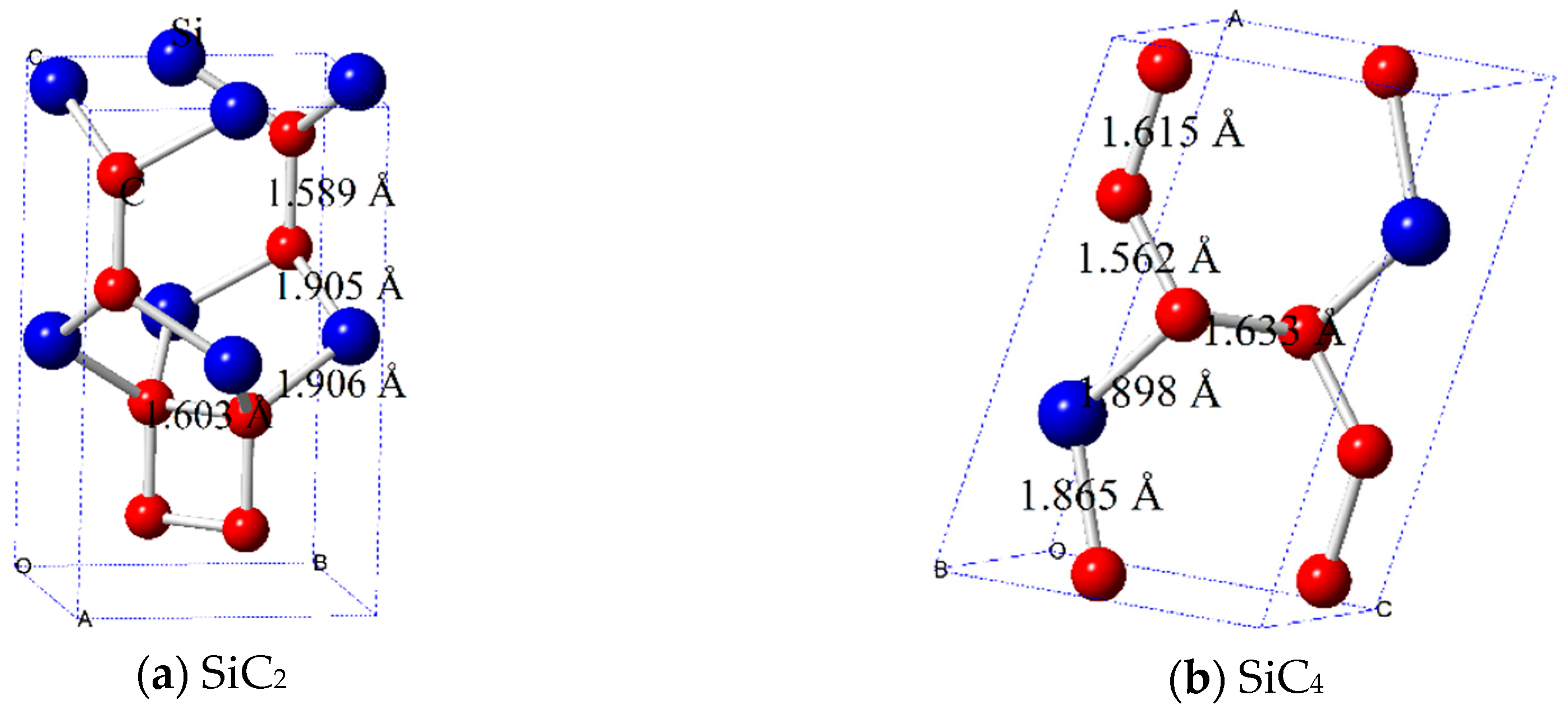{kind=link}
{kind=link}
{kind=link}
{kind=link}
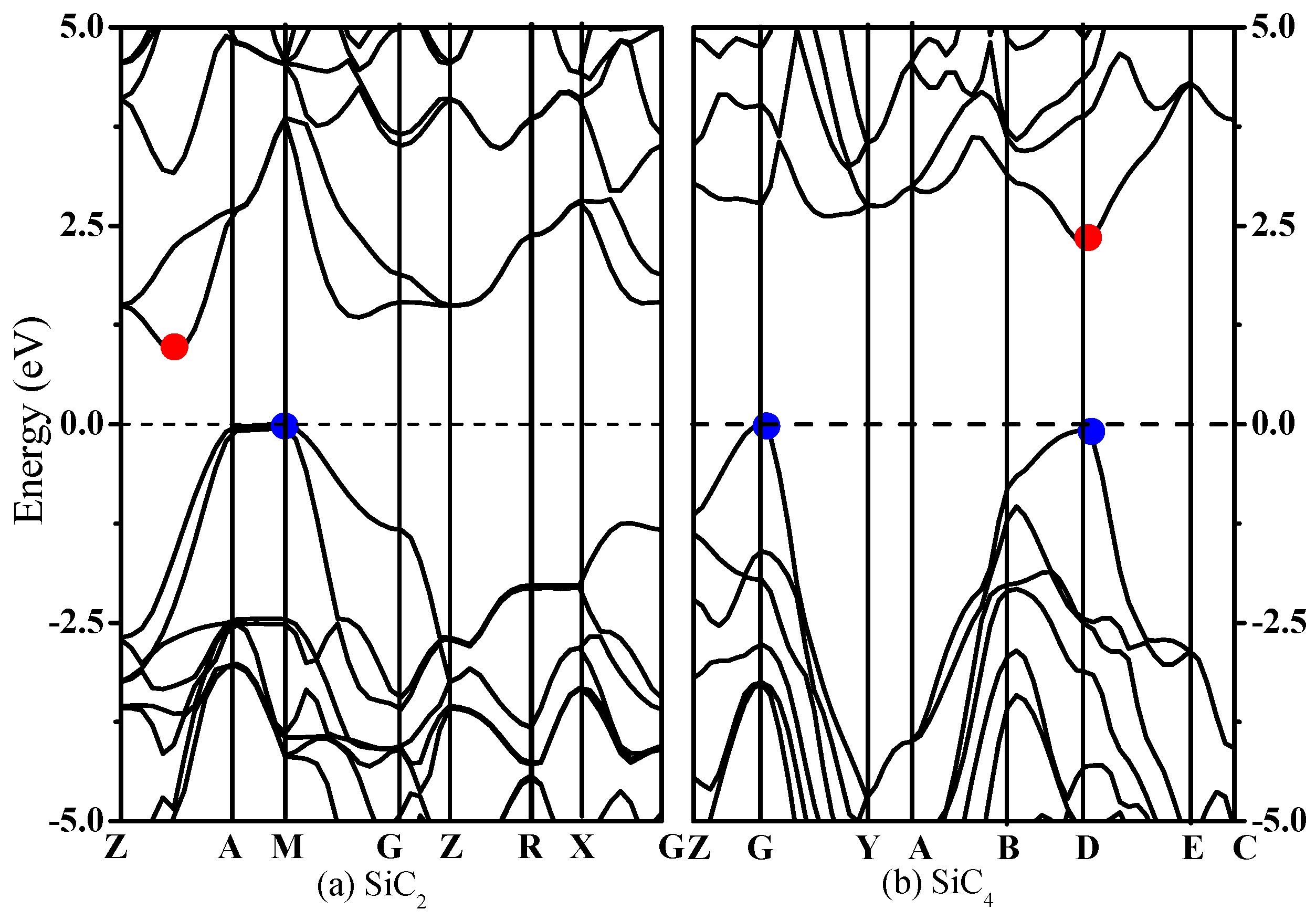{kind=link}
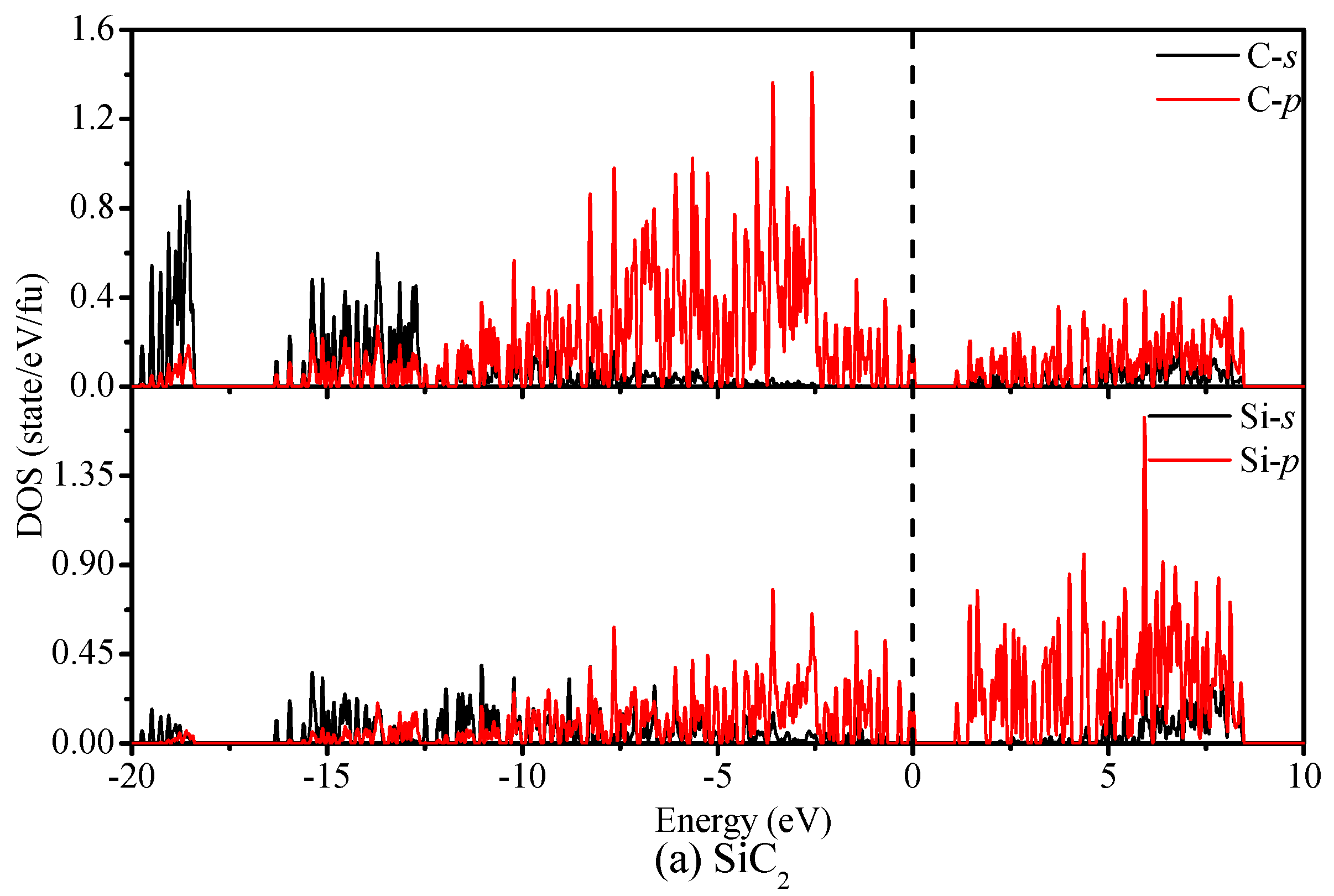{kind=link}
{kind=link}
| Materials | SG | Methods | a | b | C | β | B | G | E | v |
|---|---|---|---|---|---|---|---|---|---|---|
| SiC2 | P42nm | PBE 1 | 4.197 | 7.107 | 203 | 162 | 384 | 0.18 | ||
| PBEsol 1 | 4.193 | 7.100 | 205 | 172 | 403 | 0.17 | ||||
| CA-PZ 1 | 4.141 | 7.010 | 217 | 178 | 419 | 0.18 | ||||
| SiC4 | P21/m | PBE 1 | 6.755 | 2.763 | 4.379 | 75.78 | 285 | 258 | 595 | 0.15 |
| PBEsol 1 | 6.744 | 2.749 | 4.369 | 75.75 | 230 | 254 | 557 | 0.10 | ||
| CA-PZ 1 | 6.752 | 2.762 | 4.378 | 75.81 | 250 | 274 | 602 | 0.10 | ||
| SiC | F-43m | PBE 1 | 4.348 | 217 | 187 | 436 | 0.17 | |||
| PBEsol 1 | 4.362 | 216 | 186 | 433 | 0.17 | |||||
| CA-PZ 1 | 4.300 | 229 | 200 | 465 | 0.16 | |||||
| PBE 2 | 4.380 | 235 5 | ||||||||
| PBE 3 | 4.344 | 224 6 | ||||||||
| Exp. 4 | 4.360 | 227 7 | 192 | 448 | 0.17 |
| Materials | Methods | C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 | C15 | C25 | C35 | C46 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SiC2 | PBE 1 | 373 | 447 | 172 | 181 | 94 | 114 | |||||||
| PBEsol 1 | 398 | 449 | 186 | 177 | 103 | 100 | ||||||||
| CA-PZ 1 | 409 | 483 | 191 | 191 | 101 | 115 | ||||||||
| SiC4 | PBE 1 | 606 | 650 | 648 | 316 | 280 | 196 | 58 | 188 | 87 | −7 | −9 | −22 | −19 |
| PBEsol 1 | 576 | 560 | 619 | 290 | 285 | 187 | 65 | 117 | 42 | −16 | 3 | −6 | −11 | |
| CA-PZ 1 | 609 | 612 | 677 | 313 | 305 | 203 | 59 | 121 | 54 | −23 | −1 | −8 | −15 | |
| SiC | PBE 1 | 385 | 243 | 132 | ||||||||||
| PBEsol 1 | 381 | 244 | 133 | |||||||||||
| CA-PZ 1 | 408 | 261 | 140 | |||||||||||
| PBE 2 | 382 | 239 | 128 | |||||||||||
| CA-PZ 3 | 390 | 253 | 134 | |||||||||||
| Exp. 4 | 390 | 256 | 142 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Chai, C.; Wei, Q.; Yang, Y. The Mechanical and Electronic Properties of Carbon-Rich Silicon Carbide. Materials 2016, 9, 333. https://doi.org/10.3390/ma9050333
Fan Q, Chai C, Wei Q, Yang Y. The Mechanical and Electronic Properties of Carbon-Rich Silicon Carbide. Materials. 2016; 9(5):333. https://doi.org/10.3390/ma9050333
Chicago/Turabian StyleFan, Qingyang, Changchun Chai, Qun Wei, and Yintang Yang. 2016. "The Mechanical and Electronic Properties of Carbon-Rich Silicon Carbide" Materials 9, no. 5: 333. https://doi.org/10.3390/ma9050333
APA StyleFan, Q., Chai, C., Wei, Q., & Yang, Y. (2016). The Mechanical and Electronic Properties of Carbon-Rich Silicon Carbide. Materials, 9(5), 333. https://doi.org/10.3390/ma9050333