
Quantitative and Qualitative Analysis of Surface Modified Cellulose Utilizing TGA-MS

Abstract
:1. Introduction
2. Results and Discussion
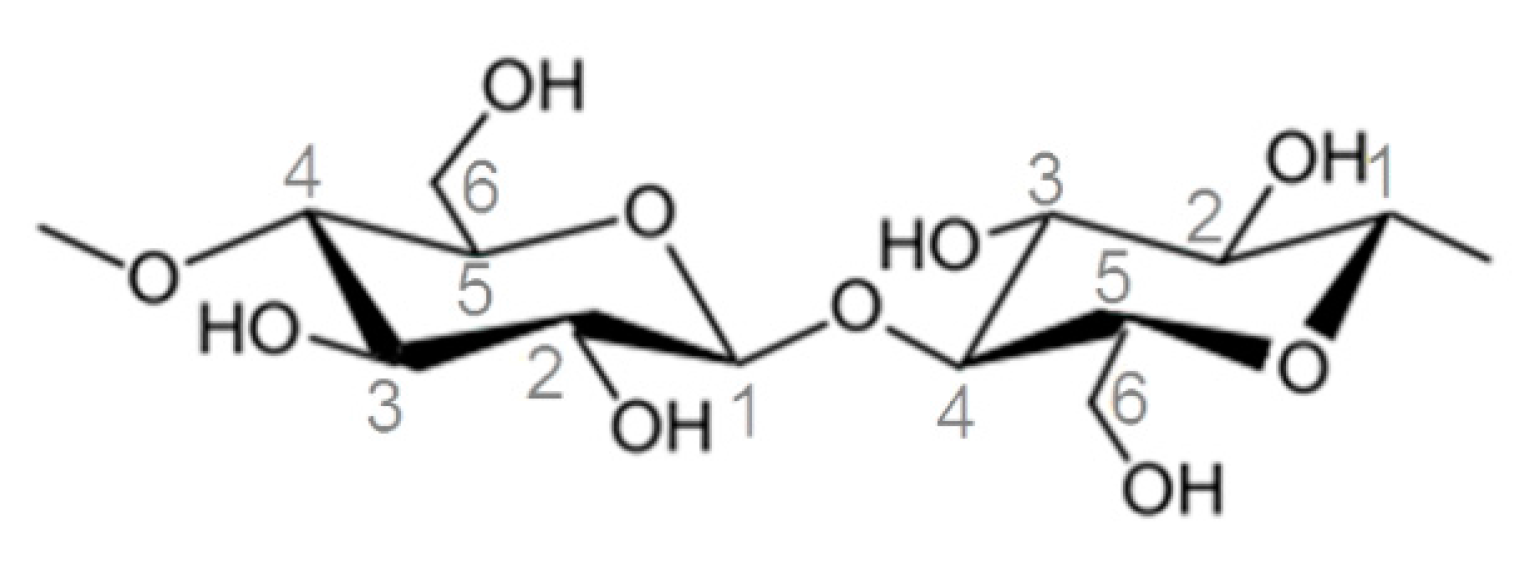
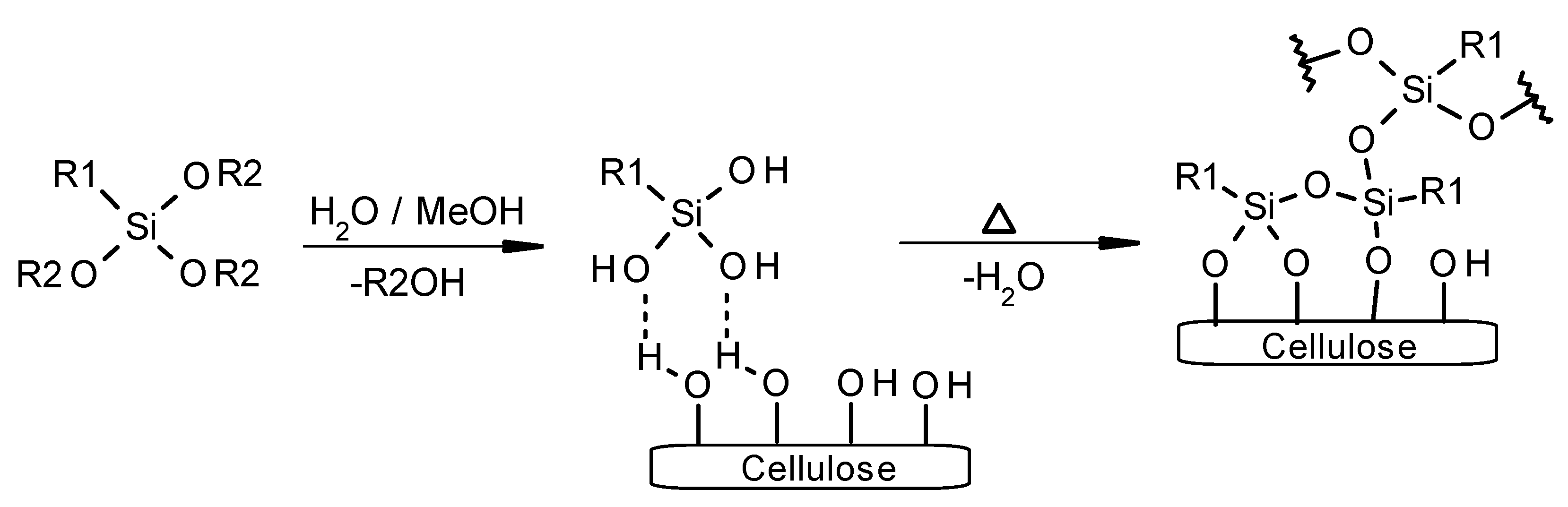
2.1. Surface Modification of Microcrystalline Cellulose
2.2. Characterization of Surface Modified Cellulose
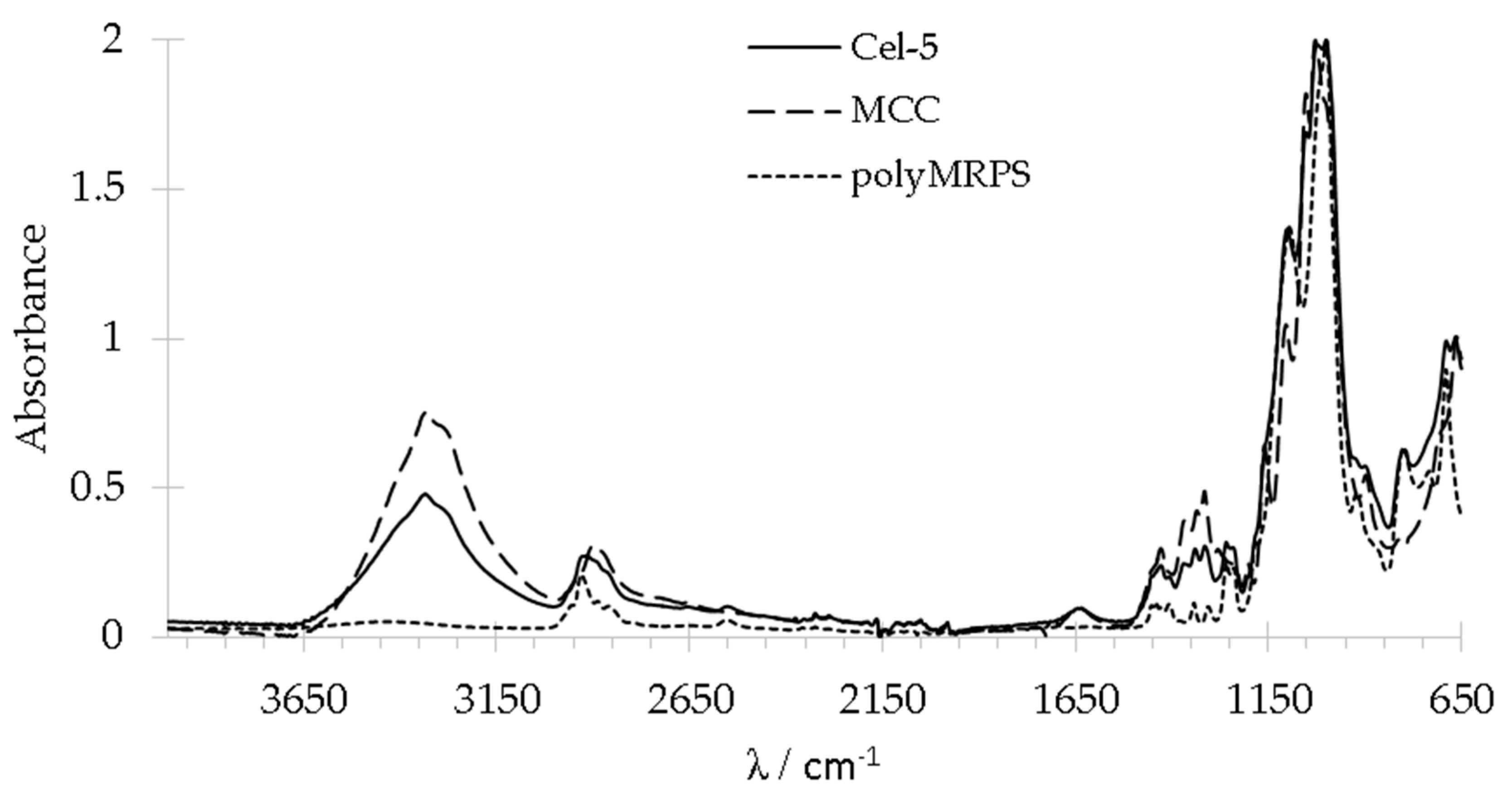
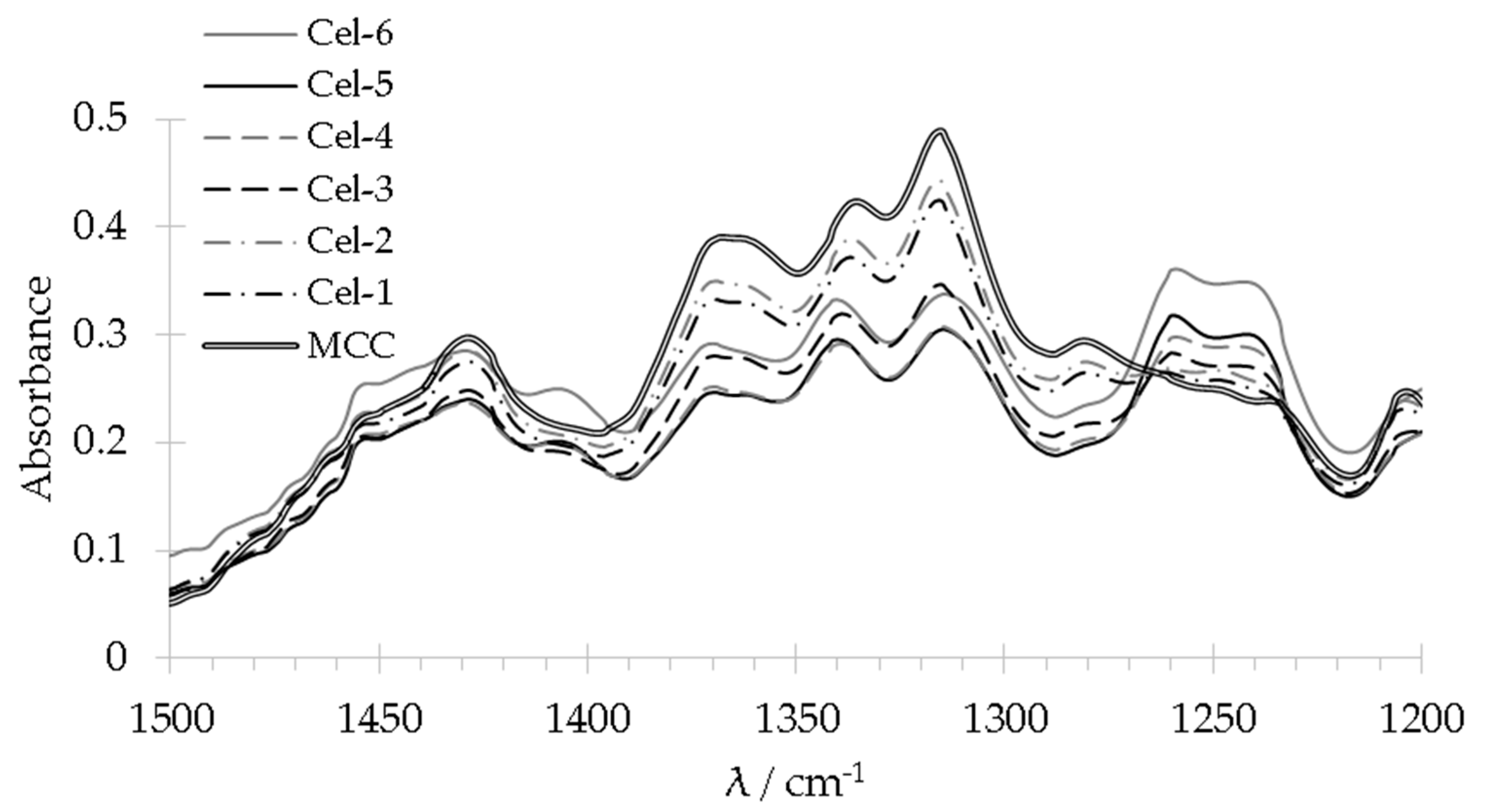
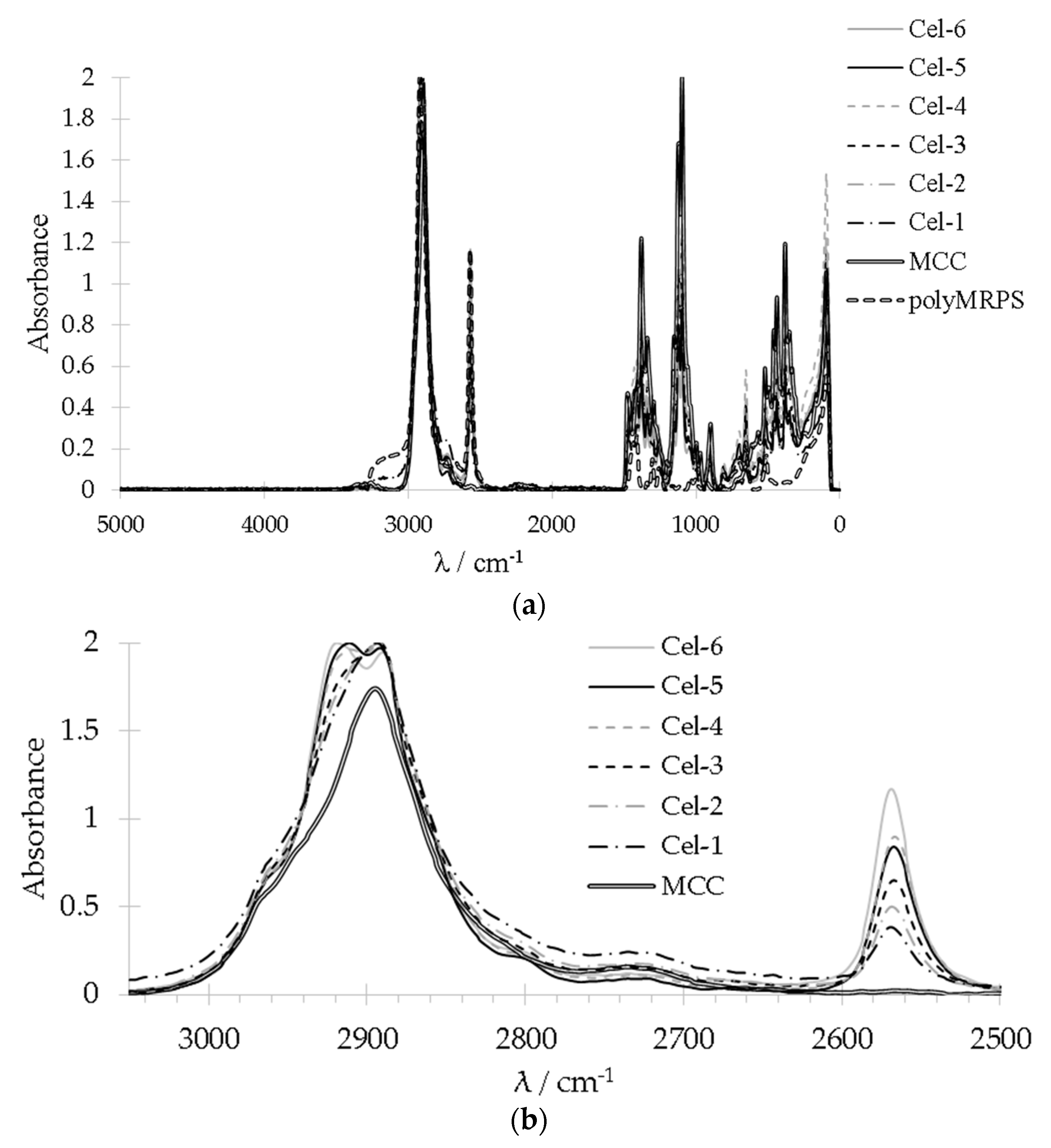
2.2.1. Analysis of Functional Groups by FT-IR and Raman Spectroscopy
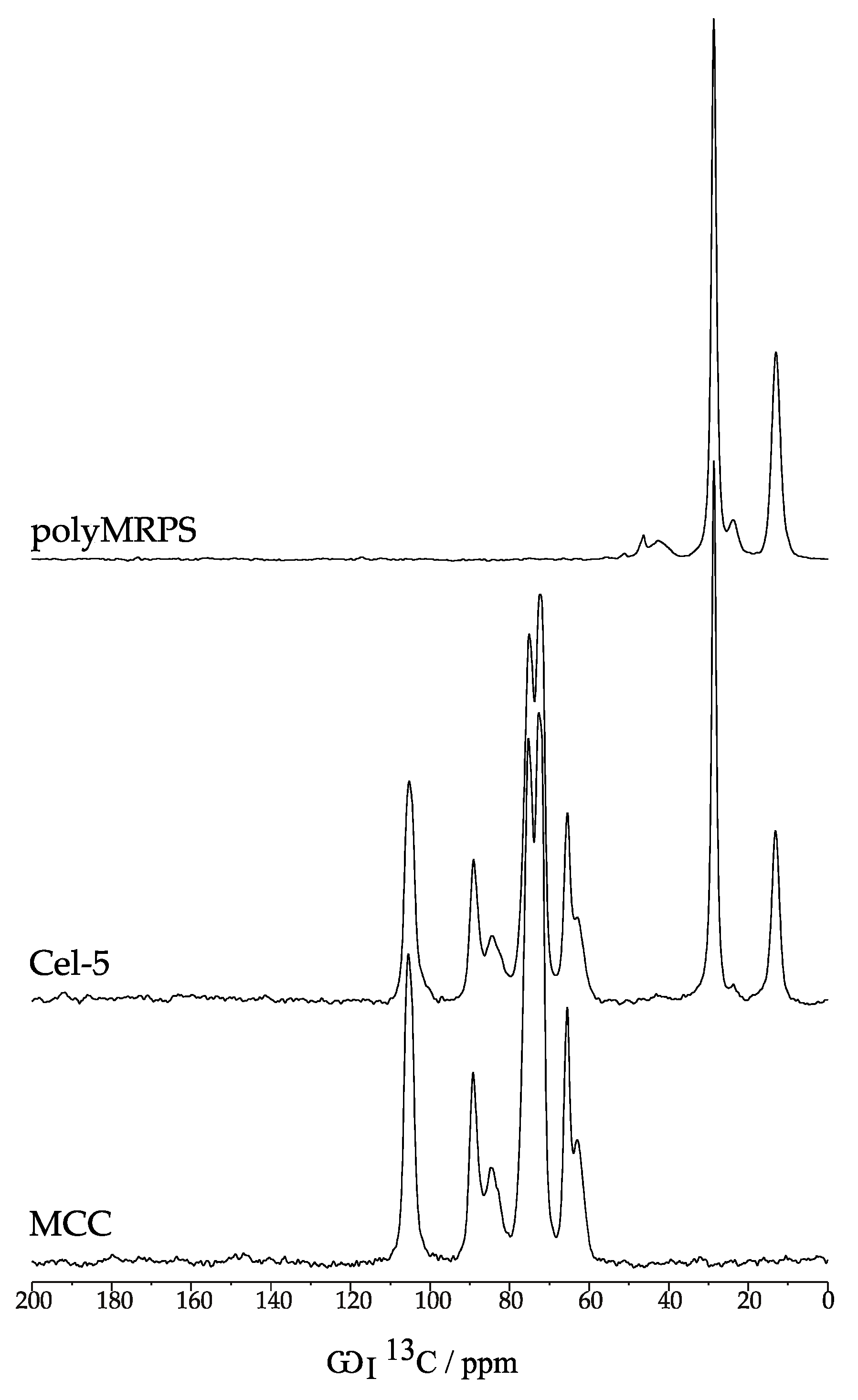
2.2.2. Morphology and Binding State
2.3. Thermal Properties and Silane Loading Determination
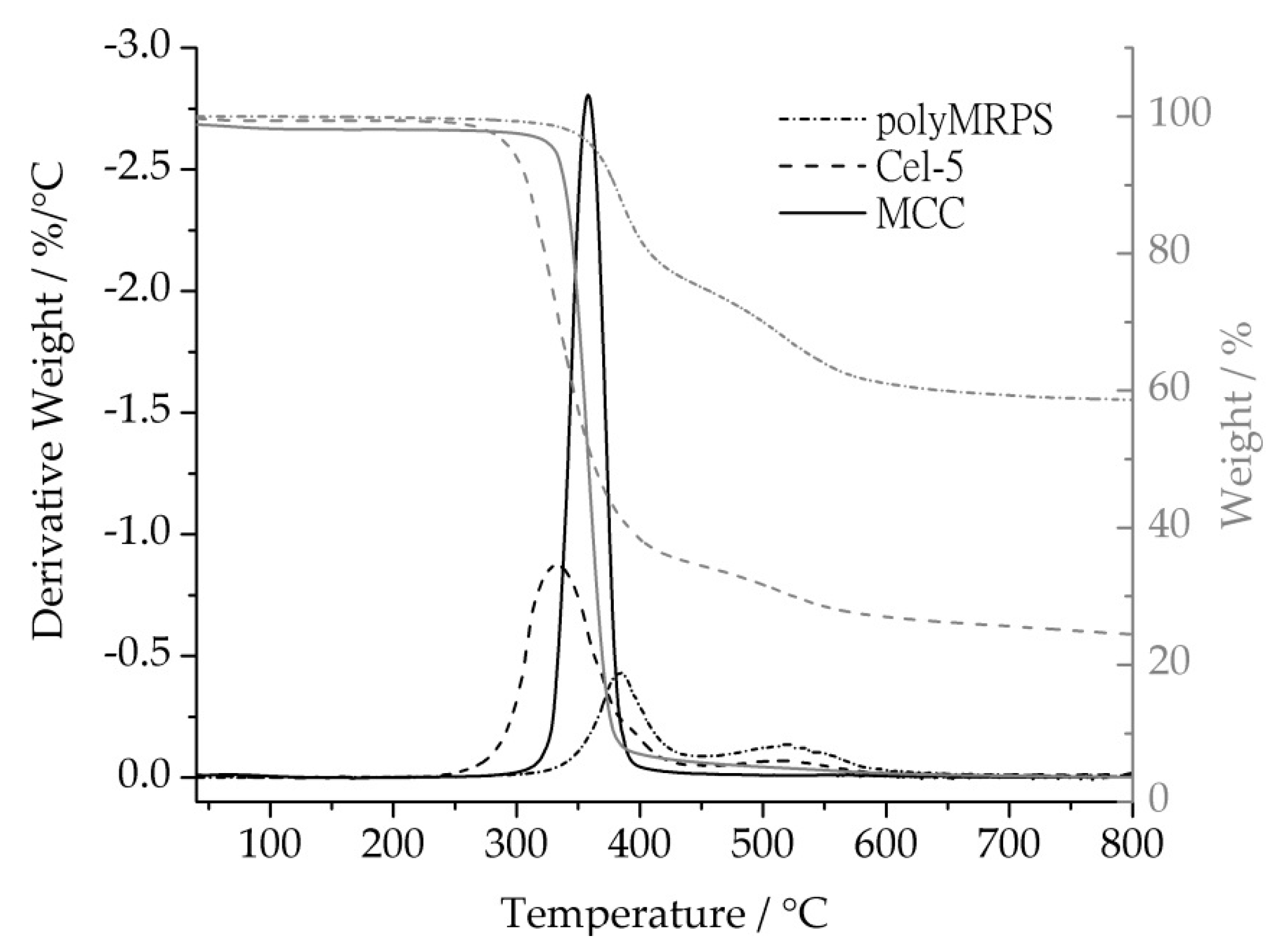
2.3.1. Studying Thermal Behavior with Thermogravimetric Analyses
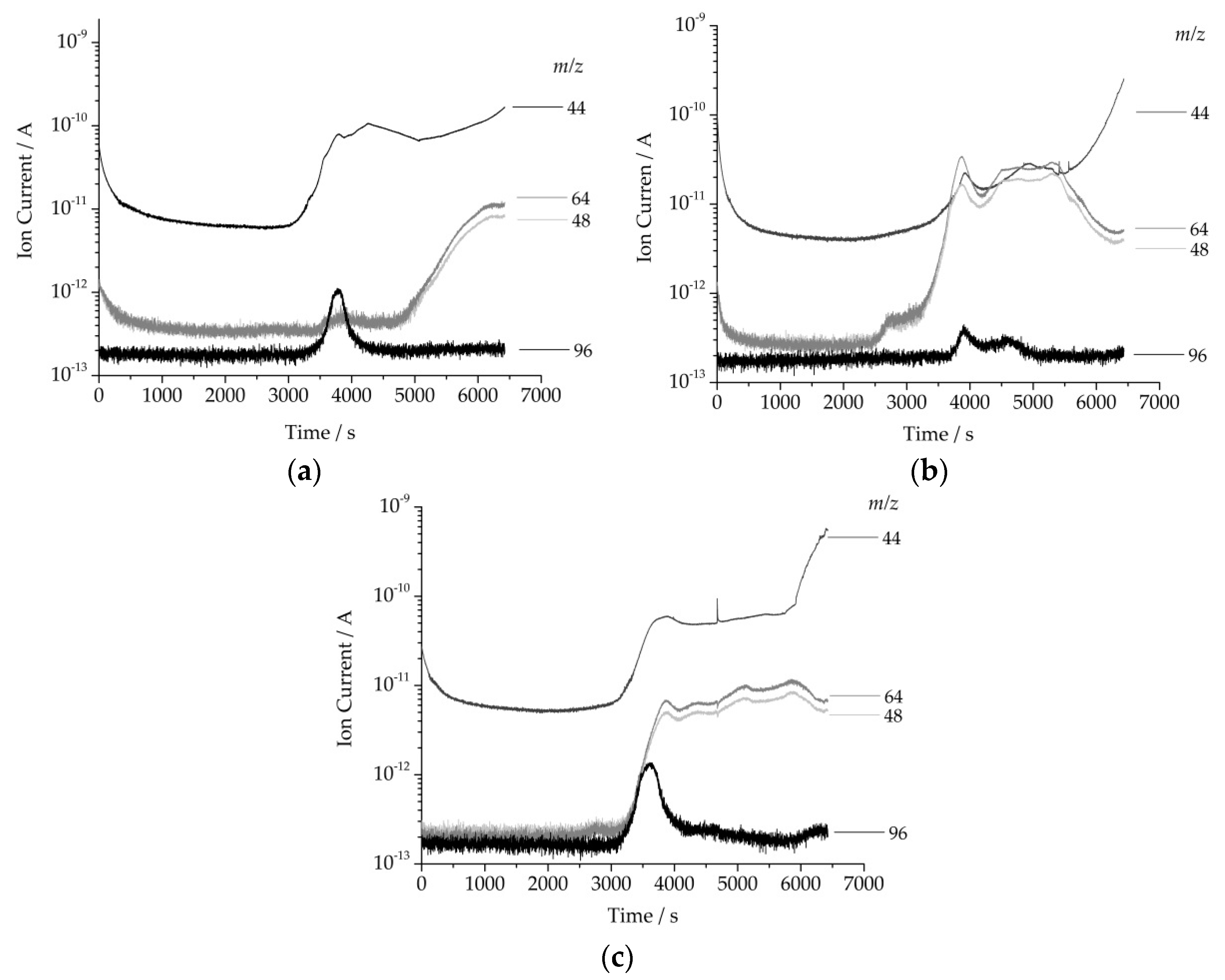
2.3.2. Anaylsis of Evolved Gas During Heating by MS
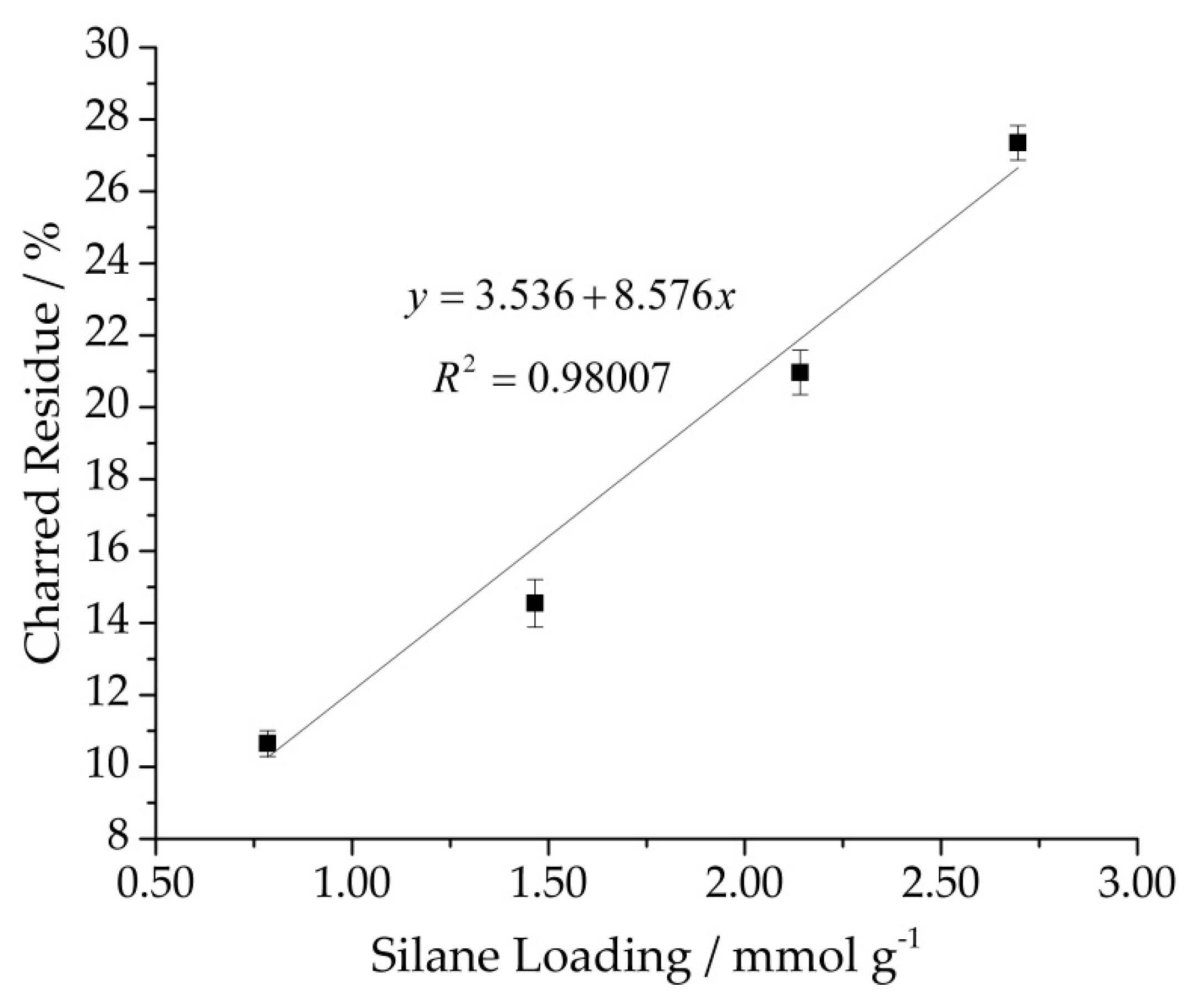
2.3.3. Determination of Silane Loading by TGA and EA Correlation
3. Materials and Methods
3.1. Characterization
3.1.1. Fourier Transformed Infrared Spectroscopy (FT-IR)
3.1.2. Raman Spectroscopy
3.1.3. Elemental Analysis
3.1.4. Thermogravimetric Analysis Coupled with a Quadrupol Mass Spectrometer (TGA-MS)
3.1.5. Solid State CP-MAS 13C NMR Spectroscopy
3.1.6. Polarized Light Optical Microscopy
3.2. Chemical Modification
Silylation of Microcrystalline Cellulose
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| EA | Elemental analysis |
| CP-MAS | Cross polarization magic angle spinning |
| FT-IR | Fourier transform infrared spectroscopy |
| MCC | Microcrystalline cellulose |
| MID | Multi ion detector |
| MRPS | (3–mercaptopropyl)trimethoxysilane |
| RF | Radio frequency |
| SPINAL 64 | Small phase incremental alternation with 64 steps |
| TGA-MS | Thermogravimetric analysis coupled with a mass spectrometer |
References
- Ku, H.; Wang, H.; Pattarachaiyakoop, N.; Trada, M. A review on the tensile properties of natural fiber reinforced polymer composites. Compos. Part B 2011, 42, 856–873. [Google Scholar] [CrossRef] [Green Version]
- Koronis, G.; Silva, A.; Fontul, M. Green composites: A review of adequate materials for automotive applications. Compos. Part B 2013, 44, 120–127. [Google Scholar] [CrossRef]
- Herrera-Franco, P.; Valadez-González, A. Mechanical properties of continuous natural fibre-reinforced polymer composites. Compos. Part A 2004, 35, 339–345. [Google Scholar] [CrossRef]
- Wambua, P.; Ivens, J.; Verpoest, I. Natural fibres: Can they replace glass in fibre reinforced plastics? Compos. Sci. Technol. 2003, 63, 1259–1264. [Google Scholar] [CrossRef]
- Qiu, X.; Hu, S. “Smart” Materials Based on Cellulose: A Review of the Preparations, Properties, and Applications. Materials 2013, 6, 738–781. [Google Scholar] [CrossRef]
- Natarajan, T.; Kumaravel, A.; Palanivelu, R. Extraction and characterization of natural cellulosic fiber from Passiflora foetida stem. Int. J. Polym. Anal. Charact. 2016. [Google Scholar] [CrossRef]
- Koschek, K. Design of natural fiber composites utilizing interfacial crystallinity and affinity. Compos. Part A 2015, 69, 21–29. [Google Scholar] [CrossRef]
- Sawpan, M.A.; Pickering, K.L.; Fernyhough, A. Effect of fibre treatments on interfacial shear strength of hemp fibre reinforced polylactide and unsaturated polyester composites. Compos. Part A 2011, 42, 1189–1196. [Google Scholar] [CrossRef]
- Uehara, T.; Sakata, I. Effect of corona discharge treatment on cellulose prepared from beech wood. J. Appl. Polym. Sci. 1990, 41, 1695–1706. [Google Scholar] [CrossRef]
- Kolářová, K.; Vosmanská, V.; Rimpelová, S.; Švorčík, V. Effect of plasma treatment on cellulose fiber. Cellulose 2013, 20, 953–961. [Google Scholar] [CrossRef]
- Miao, C.; Hamad, W.Y. Cellulose reinforced polymer composites and nanocomposites: A critical review. Cellulose 2013, 20, 2221–2262. [Google Scholar] [CrossRef]
- Kabir, M.M.; Wang, H.; Lau, K.T.; Cardona, F. Chemical treatments on plant-based natural fibre reinforced polymer composites: An overview. Compos. Part B 2012, 43, 2883–2892. [Google Scholar] [CrossRef]
- Keener, T.; Stuart, R.; Brown, T. Maleated coupling agents for natural fibre composites. Compos. Part A 2004, 35, 357–362. [Google Scholar] [CrossRef]
- Timhadjelt, L.; Serier, A.; Belgacem, M.N.; Bras, J. Elaboration of cellulose based nanobiocomposite: Effect of cellulose nanocrystals surface treatment and interface “melting”. Ind. Crop. Prod. 2015, 72, 7–15. [Google Scholar] [CrossRef]
- Varshney, V.K.; Naithani, S. Chemical Functionalization of Cellulose Derived from Nonconventional Sources; Springer-Verlag: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Zhou, J.; Li, Q.; Song, Y.; Zhang, L.; Lin, X. A facile method for the homogeneous synthesis of cyanoethyl cellulose in NaOH/urea aqueous solutions. Polym. Chem. 2010, 1, 1662–1668. [Google Scholar] [CrossRef]
- Missoum, K.; Belgacem, M.; Bras, J. Nanofibrillated Cellulose Surface Modification: A Review. Materials 2013, 6, 1745–1766. [Google Scholar] [CrossRef]
- Rajkumar, R.; Manikandan, A.; Saravanakumar, S.S. Physicochemical properties of alkali-treated new cellulosic fiber from cotton shell. Int. J. Polym. Anal. Charact. 2016, 21, 359–364. [Google Scholar] [CrossRef]
- Hettegger, H.; Sumerskii, I.; Sortino, S.; Potthast, A.; Rosenau, T. Silane Meets Click Chemistry: Towards the Functionalization of Wet Bacterial Cellulose Sheets. ChemSusChem 2015, 8, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Rachini, A.; Le Troedec, M.; Peyratout, C.; Smith, A. Chemical modification of hemp fibers by silane coupling agents. J. Appl. Polym. Sci. 2012, 123, 601–610. [Google Scholar] [CrossRef]
- Rojo, E.; Alonso, M.V.; Del Saz-Orozco, B.; Oliet, M.; Rodriguez, F. Optimization of the silane treatment of cellulosic fibers from eucalyptus wood using response surface methodology. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Xie, Y.; Hill, C.A.; Xiao, Z.; Militz, H.; Mai, C. Silane coupling agents used for natural fiber/polymer composites: A review. Compos. Part A 2010, 41, 806–819. [Google Scholar] [CrossRef]
- Thakur, V.K.; Singha, A.S.; Kaur, I.; Nagarajarao, R.P.; Liping, Y. Silane Functionalization of Saccaharum cilliare Fibers: Thermal, Morphological, and Physicochemical Study. Int. J. Polym. Anal. Charact. 2010, 15, 397–414. [Google Scholar] [CrossRef]
- Singha, A.S.; Thakur, V.K. Synthesis and Characterizations of Silane Treated Grewia optiva Fibers. Int. J. Polym. Anal. Charact. 2009, 14, 301–321. [Google Scholar] [CrossRef]
- Singha, A.S.; Thakur, V.K.; Mehta, I.K.; Shama, A.; Khanna, A.J.; Rana, R.K.; Rana, A.K. Surface-Modified Hibiscus sabdariffa Fibers: Physicochemical, Thermal, and Morphological Properties Evaluation. Int. J. Polym. Anal. Charact. 2009, 14, 695–711. [Google Scholar] [CrossRef]
- Rachini, A.; Le Troedec, M.; Peyratout, C.; Smith, A. Comparison of the thermal degradation of natural, alkali-treated and silane-treated hemp fibers under air and an inert atmosphere. J. Appl. Polym. Sci. 2009, 112, 226–234. [Google Scholar] [CrossRef]
- Ly, B.; Belgacem, M.N.; Bras, J.; Brochier Salon, M.C. Grafting of cellulose by fluorine-bearing silane coupling agents. Mater. Sci. Eng. C 2010, 30, 343–347. [Google Scholar] [CrossRef]
- Eklund, T.; Britcher, L.; Bäckman, J.; Rosenholm, J.B. Thermogravimetric Analysis of Aminopropyl-trimethoxysilane Adsorbed on Silica Support. J. Therm. Anal. Calorim. 1999, 58, 67–76. [Google Scholar] [CrossRef]
- Cestari, A.R.; Airoldi, C. A new elemental analysis method based on thermogravimetric data and applied to alkoxysilane immobilized on silicas. J. Therm. Anal. 1995, 44, 79–87. [Google Scholar] [CrossRef]
- Wilson, A.M.; Zank, G.; Eguchi, K.; Xing, W.; Yates, B.; Dahn, J.R. Polysiloxane Pyrolysis. Chem. Mater. 1997, 9, 1601–1606. [Google Scholar] [CrossRef]
- Brochier Salon, M.-C.; Abdelmouleh, M.; Boufi, S.; Belgacem, M.N.; Gandini, A. Silane adsorption onto cellulose fibers: Hydrolysis and condensation reactions. J. Colloid Interface Sci. 2005, 289, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Abdelmouleh, M.; Boufi, S.; ben Salah, A.; Belgacem, M.N.; Gandini, A. Interaction of Silane Coupling Agents with Cellulose. Langmuir 2002, 18, 3203–3208. [Google Scholar] [CrossRef]
- Salon, M.-C.B.; Gerbaud, G.; Abdelmouleh, M.; Bruzzese, C.; Boufi, S.; Belgacem, M.N. Studies of interactions between silane coupling agents and cellulose fibers with liquid and solid-state NMR. Magn. Reson. Chem. 2007, 45, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Baker, J.O.; Himmel, M.E.; Parilla, P.A.; Johnson, D.K. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Hesse-Ertelt, S.; Witter, R.; Ulrich, A.S.; Kondo, T.; Heinze, T. Spectral assignments and anisotropy data of cellulose Iα: 13C NMR chemical shift data of cellulose Iα determined by INADEQUATE and RAI techniques applied to uniformly 13C-labeled bacterial celluloses of different Gluconacetobacter xylinus strains. Magn. Reson. Chem. 2008, 46, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Semsarilar, M.; Guthrie, J.T.; Perrier, S. Cellulose modification by polymer grafting: A review. Chem. Soc. Rev. 2009, 38, 2046–2064. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Cho, J.; Tompsett, G.A.; Westmoreland, P.R.; Huber, G.W. Kinetics and Mechanism of Cellulose Pyrolysis. J. Phys. Chem. C 2009, 113, 20097–20107. [Google Scholar] [CrossRef]
- Xu, L.; Yang, J.; Li, Y.; Liu, Z. Behavior of organic sulfur model compounds in pyrolysis under coal-like environment. Fuel Process. Technol. 2004, 85, 1013–1024. [Google Scholar] [CrossRef]
- Pines, A. Proton-Enhanced Nuclear Induction Spectroscopy. A Method for High Resolution NMR of Dilute Spins in Solids. J. Chem. Phys. 1972, 56, 1776–1777. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Cel–1 | Cel–2 | Cel–3 | Cel–4 | Cel–5 | Cel–6 |
|---|---|---|---|---|---|---|
| [MRPS]0 (mol/L) | 0.27 | 0.54 | 0.81 | 1.08 | 1.35 | 1.62 |
| Sample | C–1 | C–4 | C–4 | C–2,3,5 | C–2,3,4 | C–6 | C–6 | –CH2–S | Si–CH2 |
|---|---|---|---|---|---|---|---|---|---|
| C | A | A | C | C | A | –CH2– | |||
| polyMRPS | – | – | – | – | – | – | – | 28.6 | 13.2 |
| Cel–5 1 | 105.4 | 89.2 | 84.5 | 75.2 | 72.6 | 65.4 | 62.9 | 28.6 | 13.2 |
| MCC | 105.4 | 89.2 | 84.5 | 75.2 | 72.6 | 65.4 | 62.9 | – | – |
| Sample | Cel–1 | Cel–2 | Cel–3 | Cel–4 | Cel–5 | Cel–6 |
|---|---|---|---|---|---|---|
| [MRPS]0 (mol/L) | 0.27 | 0.54 | 0.81 | 1.08 | 1.35 | 1.62 |
| 1 charred residue (%) | 10.7 ± 0.4 | 14.6 ± 0.7 | 21.0 ± 0.6 | 24.0 ± 0.5 | 27.4 ± 0.5 | 30.3 ± 0.8 |
| 2 MRPS loading (exp.) (mmol/g) | 0.785 | 1.466 | 2.141 | – | 2.696 | – |
| 3 MRPS loading (calc.) (mmol/g) | 0.83 ± 0.04 | 1.28 ± 0.08 | 2.03 ± 0.07 | 2.38 ± 0.05 | 2.78 ± 0.6 | 3.12 ± 0.1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loof, D.; Hiller, M.; Oschkinat, H.; Koschek, K. Quantitative and Qualitative Analysis of Surface Modified Cellulose Utilizing TGA-MS. Materials 2016, 9, 415. https://doi.org/10.3390/ma9060415
Loof D, Hiller M, Oschkinat H, Koschek K. Quantitative and Qualitative Analysis of Surface Modified Cellulose Utilizing TGA-MS. Materials. 2016; 9(6):415. https://doi.org/10.3390/ma9060415
Chicago/Turabian StyleLoof, Daniel, Matthias Hiller, Hartmut Oschkinat, and Katharina Koschek. 2016. "Quantitative and Qualitative Analysis of Surface Modified Cellulose Utilizing TGA-MS" Materials 9, no. 6: 415. https://doi.org/10.3390/ma9060415
APA StyleLoof, D., Hiller, M., Oschkinat, H., & Koschek, K. (2016). Quantitative and Qualitative Analysis of Surface Modified Cellulose Utilizing TGA-MS. Materials, 9(6), 415. https://doi.org/10.3390/ma9060415