

Polymer Models of Chromatin Imaging Data in Single Cells

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results
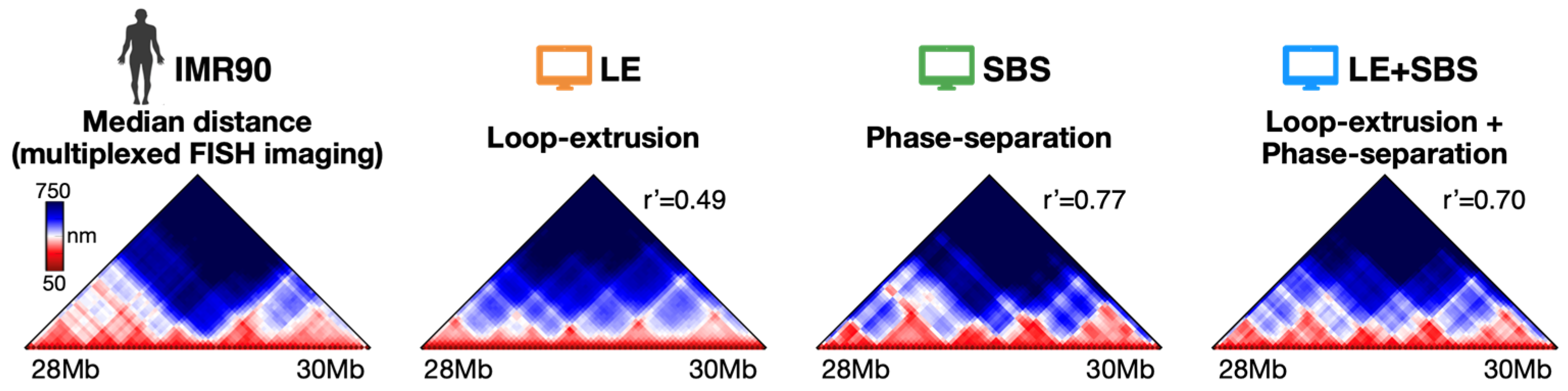
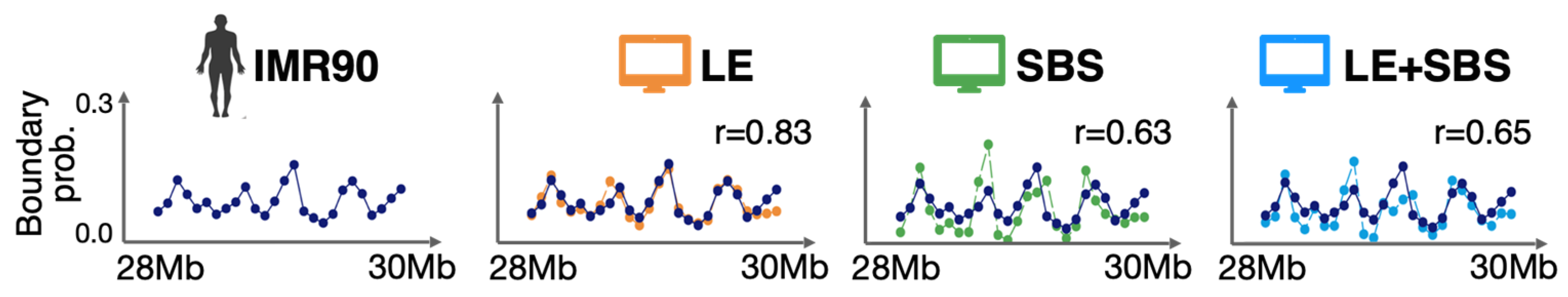
3.1. The Models Recapitulate Ensemble-Averaged Microscopy Data
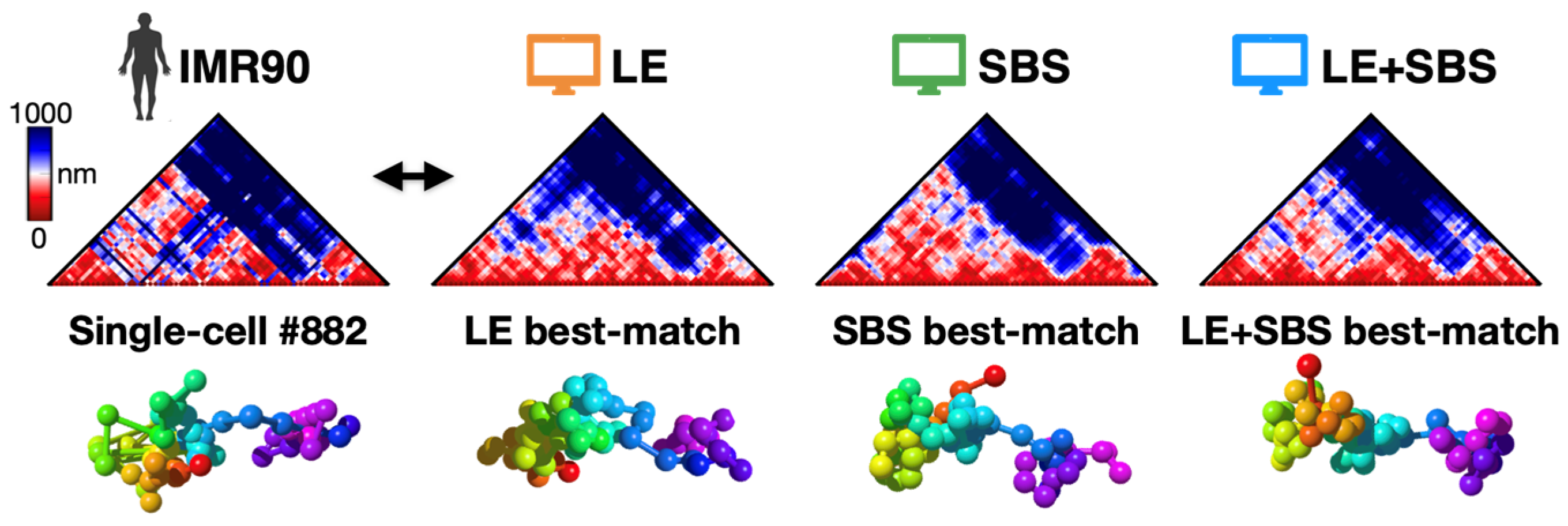
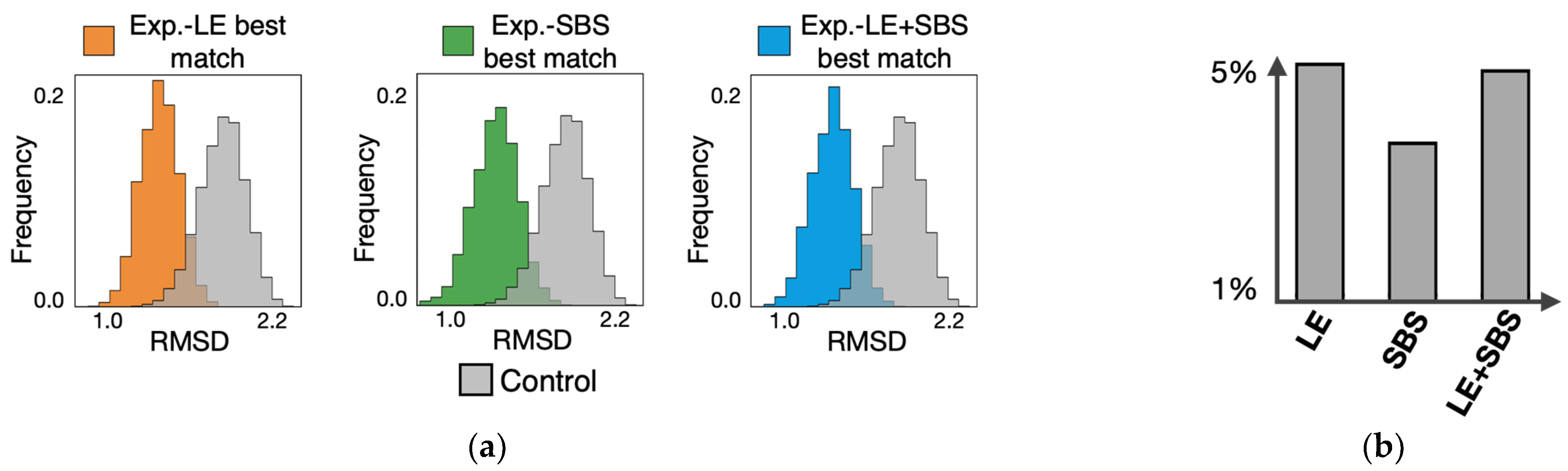
3.2. All-against-All Comparison between Single-Molecule Imaged and Model-Derived 3D Structures
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, W.A.; van Steensel, B. Genome Architecture: Domain Organization of Interphase Chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef]
- Finn, E.H.; Misteli, T. Molecular Basis and Biological Function of Variability in Spatial Genome Organization. Science 2019, 365, eaaw9498. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; Van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial Partitioning of the Regulatory Landscape of the X-Inactivation Centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Fraser, J.; Ferrai, C.; Chiariello, A.M.; Schueler, M.; Rito, T.; Laudanno, G.; Barbieri, M.; Moore, B.L.; Kraemer, D.C.; Aitken, S.; et al. Hierarchical Folding and Reorganization of Chromosomes Are Linked to Transcriptional Changes in Cellular Differentiation. Mol. Syst. Biol. 2015, 11, 852. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural Variation in the 3D Genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Bianco, S.; Lupiáñez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schöpflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer Physics Predicts the Effects of Structural Variants on Chromatin Architecture. Nat. Genet. 2018, 50, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Valton, A.-L.; Dekker, J. TAD Disruption as Oncogenic Driver. Curr. Opin. Genet. Dev. 2016, 36, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Bintu, B.; Mateo, L.J.; Su, J.-H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-Resolution Chromatin Tracing Reveals Domains and Cooperative Interactions in Single Cells. Science 2018, 362, eaau1783. [Google Scholar] [CrossRef]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-Resolution Imaging Reveals Distinct Chromatin Folding for Different Epigenetic States. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, P1502–P1515. [Google Scholar] [CrossRef]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin Extrusion Explains Key Features of Loop and Domain Formation in Wild-Type and Engineered Genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef]
- Racko, D.; Benedetti, F.; Dorier, J.; Stasiak, A. Transcription-Induced Supercoiling as the Driving Force of Chromatin Loop Extrusion during Formation of TADs in Interphase Chromosomes. Nucleic Acids Res. 2018, 46, 1648–1660. [Google Scholar] [CrossRef] [Green Version]
- Brackley, C.A.; Johnson, J.; Michieletto, D.; Morozov, A.N.; Nicodemi, M.; Cook, P.R.; Marenduzzo, D. Nonequilibrium Chromosome Looping via Molecular Slip Links. Phys. Rev. Lett. 2017, 119, 138101. [Google Scholar] [CrossRef] [PubMed]
- Banigan, E.J.; Mirny, L.A. Loop Extrusion: Theory Meets Single-Molecule Experiments. Curr. Opin. Cell Biol. 2020, 64, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Goloborodko, A.; Marko, J.F.; Mirny, L.A. Chromosome Compaction by Active Loop Extrusion. Biophys. J. 2016, 110, 2162–2168. [Google Scholar] [CrossRef]
- Goloborodko, A.; Imakaev, M.V.; Marko, J.F.; Mirny, L. Compaction and Segregation of Sister Chromatids via Active Loop Extrusion. eLife 2016, 5, e14864. [Google Scholar] [CrossRef]
- Jost, D.; Carrivain, P.; Cavalli, G.; Vaillant, C. Modeling Epigenome Folding: Formation and Dynamics of Topologically Associated Chromatin Domains. Nucleic Acids Res. 2014, 42, 9553–9561. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Fiorillo, L.; Annunziatella, C.; Esposito, A.; Musella, F.; Abraham, A.; Bianco, S.; Chiariello, A.M. Dynamic and Equilibrium Properties of Finite-Size Polymer Models of Chromosome Folding. Phys. Rev. E 2021, 104, 054402. [Google Scholar] [CrossRef]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin Organization by an Interplay of Loop Extrusion and Compartmental Segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Irani, E.; Chiariello, A.M.; Abraham, A.; Bianco, S.; Esposito, A.; Nicodemi, M. Loop-Extrusion and Polymer Phase-Separation Can Co-Exist at the Single-Molecule Level to Shape Chromatin Folding. Nat. Commun. 2022, 13, 4070. [Google Scholar] [CrossRef]
- Esposito, A.; Bianco, S.; Chiariello, A.M.; Abraham, A.; Fiorillo, L.; Conte, M.; Campanile, R.; Nicodemi, M. Polymer Physics Reveals a Combinatorial Code Linking 3D Chromatin Architecture to 1D Chromatin States. Cell Rep. 2022, 38, 110601. [Google Scholar] [CrossRef]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.-M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of Chromatin Folding Is Captured by the Strings and Binders Switch Model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef] [Green Version]
- Di Pierro, M.; Zhang, B.; Aiden, E.L.; Wolynes, P.G.; Onuchic, J.N. Transferable Model for Chromosome Architecture. Proc. Natl. Acad. Sci. USA 2016, 113, 12168–12173. [Google Scholar] [CrossRef]
- Plewczynski, D.; Kadlof, M. Computational Modelling of Three-Dimensional Genome Structure. Methods 2020, 181–182, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wolynes, P.G. Topology, Structures, and Energy Landscapes of Human Chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, A.M.; Annunziatella, C.; Bianco, S.; Esposito, A.; Nicodemi, M. Polymer Physics of Chromosome Large-Scale 3D Organisation. Sci. Rep. 2016, 6, 29775. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Brown, J.M.; Waithe, D.; Babbs, C.; Davies, J.; Hughes, J.R.; Buckle, V.J.; Marenduzzo, D. Predicting the Three-Dimensional Folding of Cis-Regulatory Regions in Mammalian Genomes Using Bioinformatic Data and Polymer Models. Genome Biol. 2016, 17, 59. [Google Scholar] [CrossRef]
- Di Stefano, M.; Paulsen, J.; Lien, T.G.; Hovig, E.; Micheletti, C. Hi-C-Constrained Physical Models of Human Chromosomes Recover Functionally-Related Properties of Genome Organization. Sci. Rep. 2016, 6, 35985. [Google Scholar] [CrossRef]
- Buckle, A.; Brackley, C.A.; Boyle, S.; Marenduzzo, D.; Gilbert, N. Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci. Mol. Cell 2018, 72, 786–797.e11. [Google Scholar] [CrossRef]
- Shi, G.; Liu, L.; Hyeon, C.; Thirumalai, D. Interphase Human Chromosome Exhibits out of Equilibrium Glassy Dynamics. Nat. Commun. 2018, 9, 3161. [Google Scholar] [CrossRef]
- Conte, M.; Fiorillo, L.; Bianco, S.; Chiariello, A.M.; Esposito, A.; Nicodemi, M. Polymer Physics Indicates Chromatin Folding Variability across Single-Cells Results from State Degeneracy in Phase Separation. Nat. Commun. 2020, 11, 3289. [Google Scholar] [CrossRef]
- Bohn, M.; Heermann, D.W. Diffusion-Driven Looping Provides a Consistent Provides a Consistent Framework for Chromatin Organization. PLoS ONE 2010, 5, e12218. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of Entangled Linear Polymer Melts: A Molecular-Dynamics Simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Rosa, A.; Everaers, R. Structure and Dynamics of Interphase Chromosomes. PLoS Comput. Biol. 2008, 4, e1000153. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Esposito, A.; Fiorillo, L.; Campanile, R.; Annunziatella, C.; Corrado, A.; Chiariello, M.G.; Bianco, S.; Chiariello, A.M. Efficient Computational Implementation of Polymer Physics Models to Explore Chromatin Structure. Int. J. Parallel, Emergent Distrib. Syst. 2019, 37, 91–102. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Anderson, J.A.; Glaser, J.; Glotzer, S.C. HOOMD-Blue: A Python Package for High-Performance Molecular Dynamics and Hard Particle Monte Carlo Simulations. Comput. Mater. Sci. 2020, 173, 109363. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D Structures of Individual Mammalian Genomes Studied by Single-Cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef]
- Theobald, D.L. Rapid Calculation of RMSDs Using a Quaternion-Based Characteristic Polynomial. Acta Crystallogr. Sect. A Found. Crystallogr. 2005, 61, 478–480. [Google Scholar] [CrossRef]
- Fiorillo, L.; Musella, F.; Conte, M.; Kempfer, R.; Chiariello, A.M.; Bianco, S.; Kukalev, A.; Irastorza-Azcarate, I.; Esposito, A.; Abraham, A.; et al. Comparison of the Hi-C, GAM and SPRITE Methods Using Polymer Models of Chromatin. Nat. Methods 2021, 18, 482–490. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stütz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-Cancer Analysis of Somatic Copy-Number Alterations Implicates IRS4 and IGF2 in Enhancer Hijacking. Nat. Genet. 2017, 49, 65–74. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, M.; Chiariello, A.M.; Abraham, A.; Bianco, S.; Esposito, A.; Nicodemi, M.; Matteuzzi, T.; Vercellone, F. Polymer Models of Chromatin Imaging Data in Single Cells. Algorithms 2022, 15, 330. https://doi.org/10.3390/a15090330
Conte M, Chiariello AM, Abraham A, Bianco S, Esposito A, Nicodemi M, Matteuzzi T, Vercellone F. Polymer Models of Chromatin Imaging Data in Single Cells. Algorithms. 2022; 15(9):330. https://doi.org/10.3390/a15090330
Chicago/Turabian StyleConte, Mattia, Andrea M. Chiariello, Alex Abraham, Simona Bianco, Andrea Esposito, Mario Nicodemi, Tommaso Matteuzzi, and Francesca Vercellone. 2022. "Polymer Models of Chromatin Imaging Data in Single Cells" Algorithms 15, no. 9: 330. https://doi.org/10.3390/a15090330
APA StyleConte, M., Chiariello, A. M., Abraham, A., Bianco, S., Esposito, A., Nicodemi, M., Matteuzzi, T., & Vercellone, F. (2022). Polymer Models of Chromatin Imaging Data in Single Cells. Algorithms, 15(9), 330. https://doi.org/10.3390/a15090330