


On the Development of Descriptor-Based Machine Learning Models for Thermodynamic Properties: Part 2—Applicability Domain and Outliers



Abstract
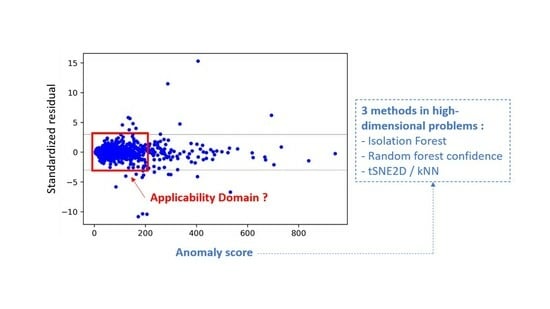
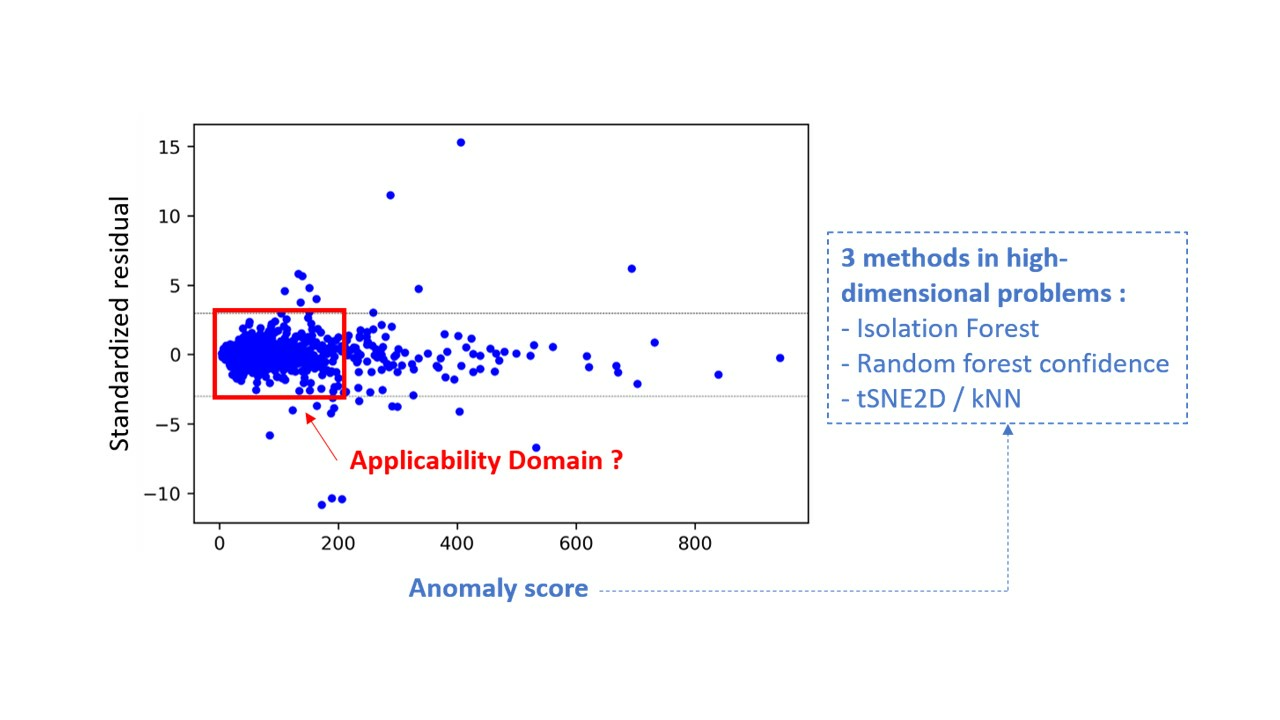
:
1. Introduction
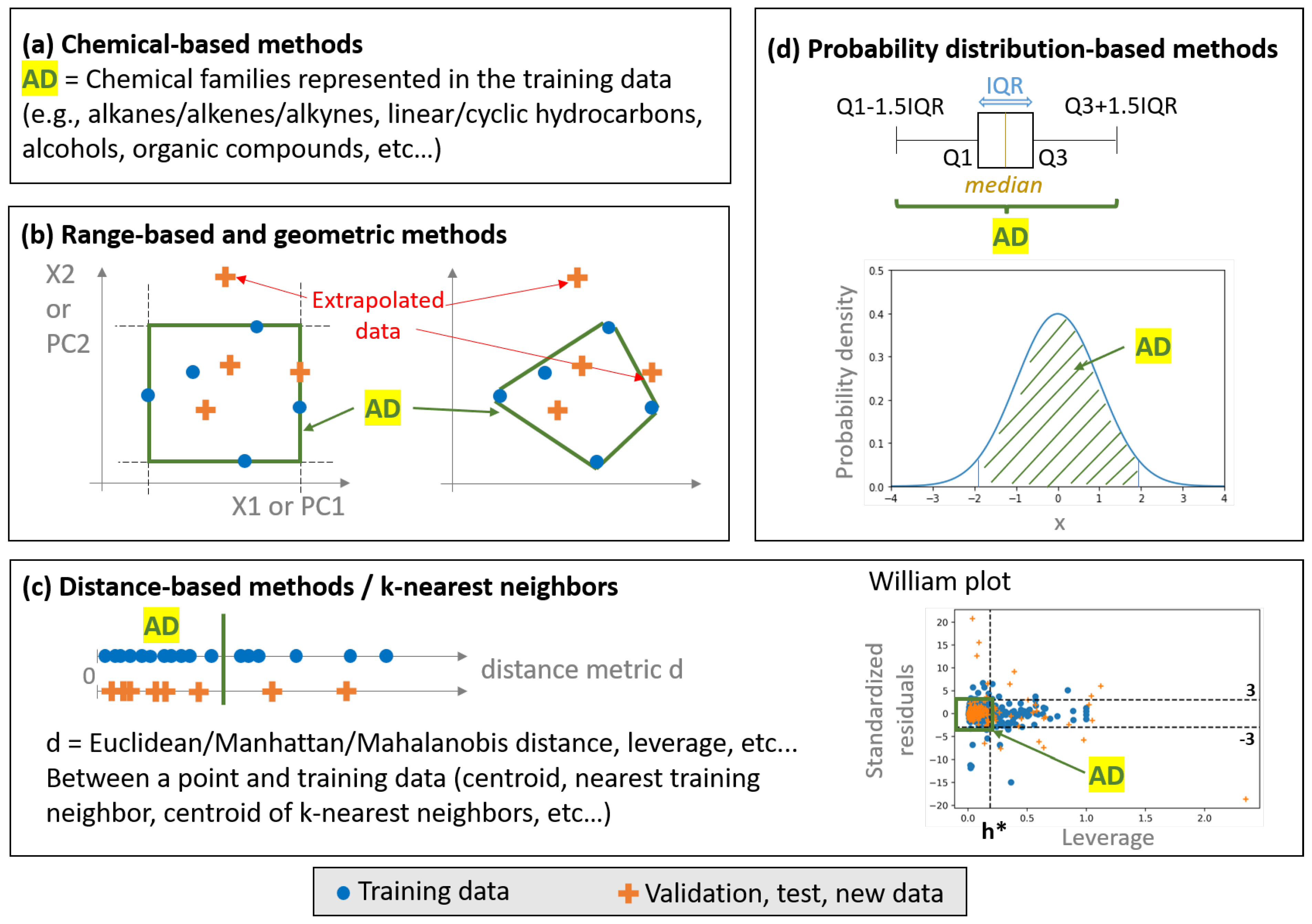
2. Overview of the Methods for AD Definition/Outlier Detection in Descriptor-Space and Their Compatibility with High-Dimensional Data
2.1. Classical Approaches
2.2. Approaches for High-Dimensional Data
3. Dataset and Methods
3.1. Dataset and ML/QSPR Models
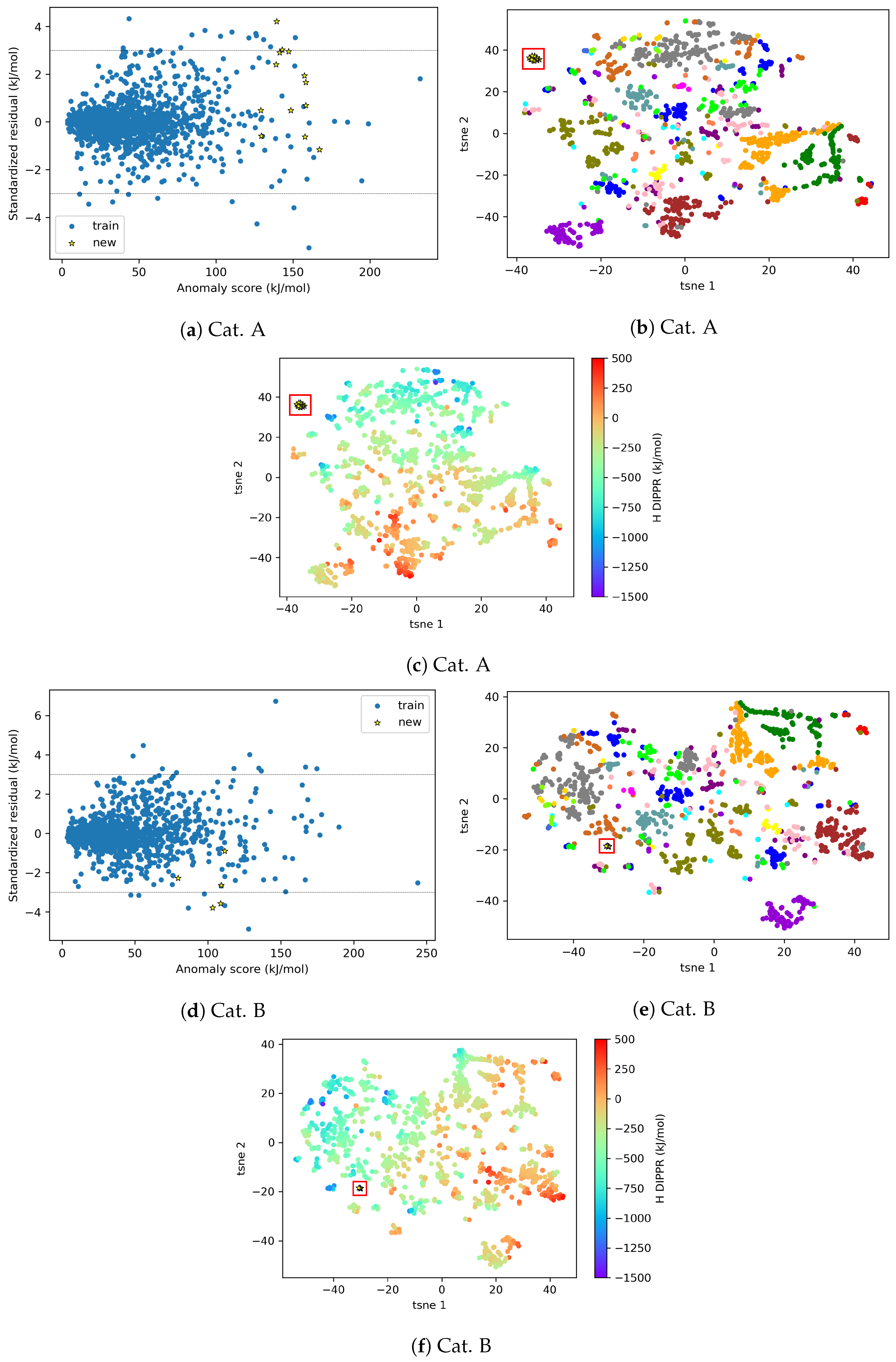
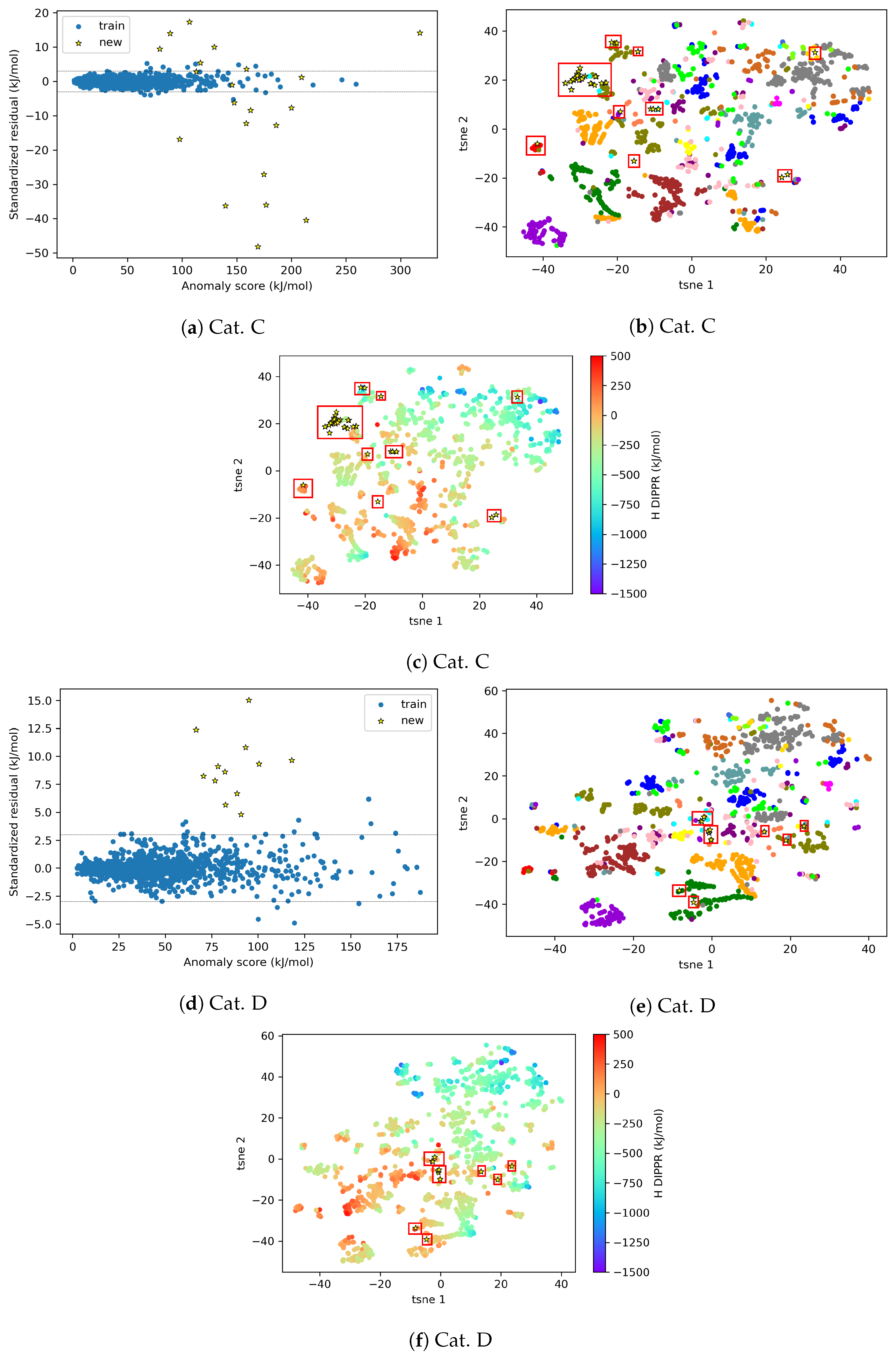
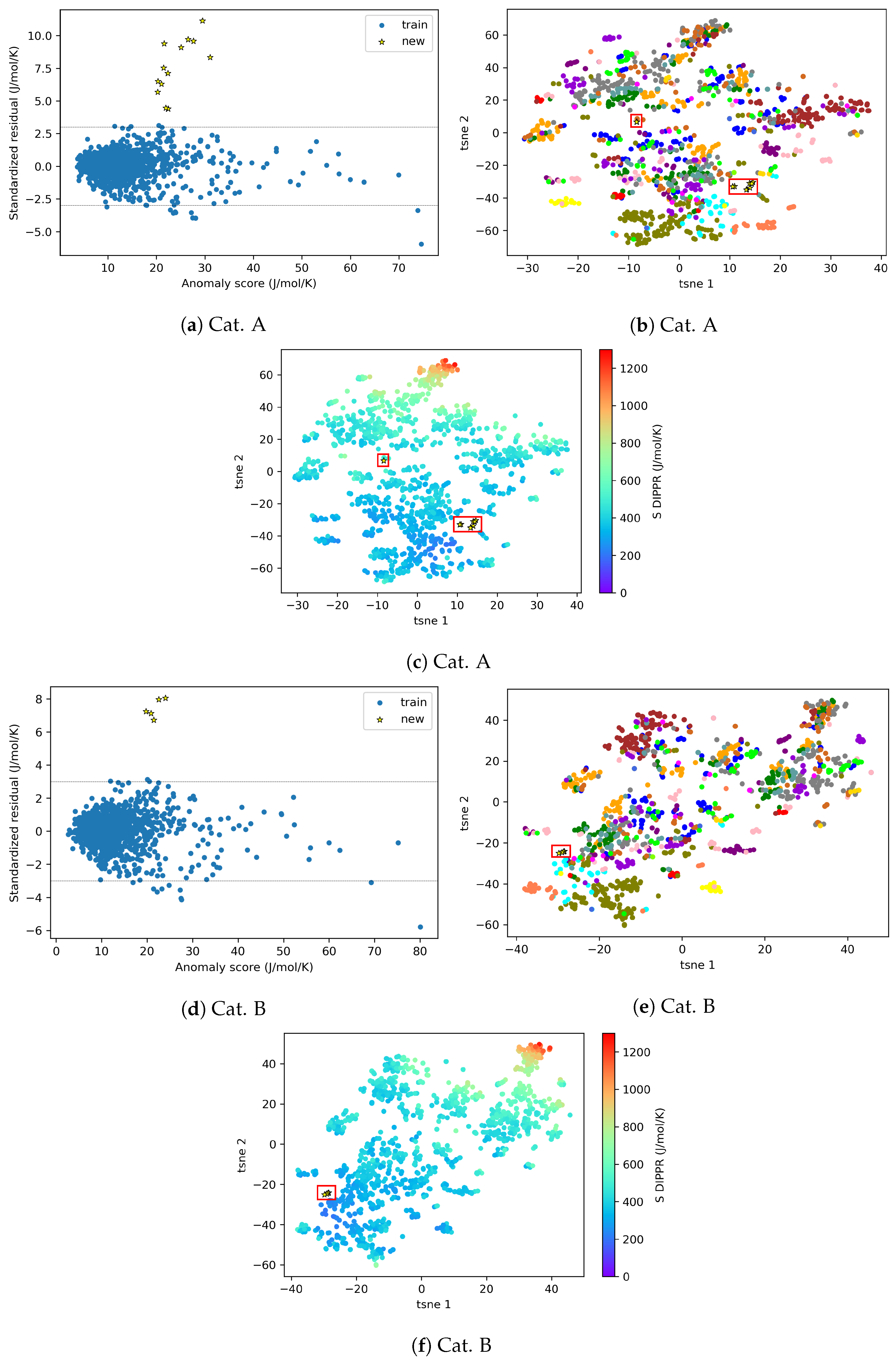
3.2. AD Visualization
3.3. AD Definition as a Data Preprocessing Method (Substudy 1)
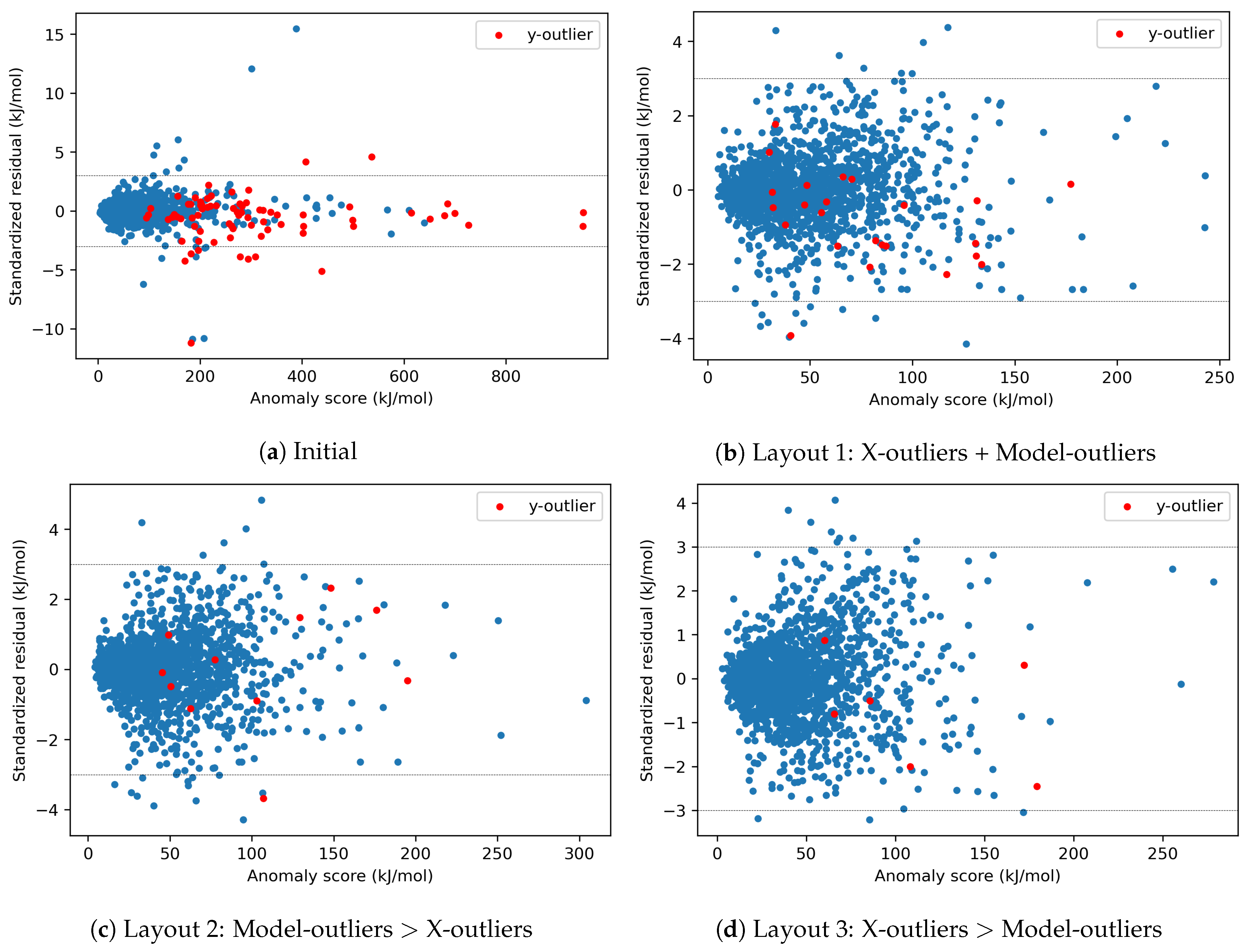
- Layout 1: simultaneous elimination of X-outliers and Model-outliers.
- Layout 2: elimination of Model-outliers followed by elimination of X-outliers.
- Layout 3: elimination of X-outliers followed by elimination of Model-outliers.
3.4. AD Definition during ML Model Construction (Substudy 2)
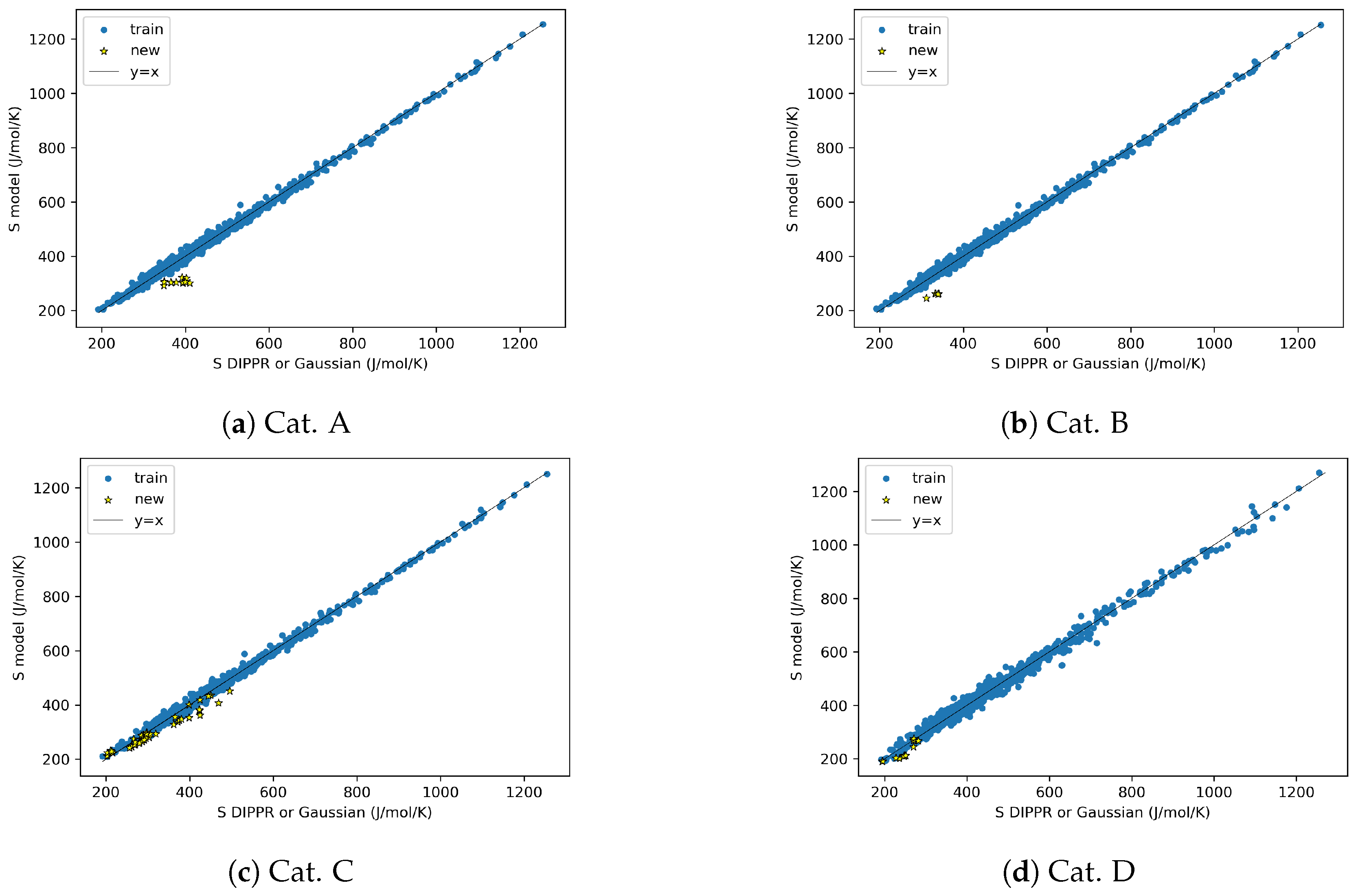
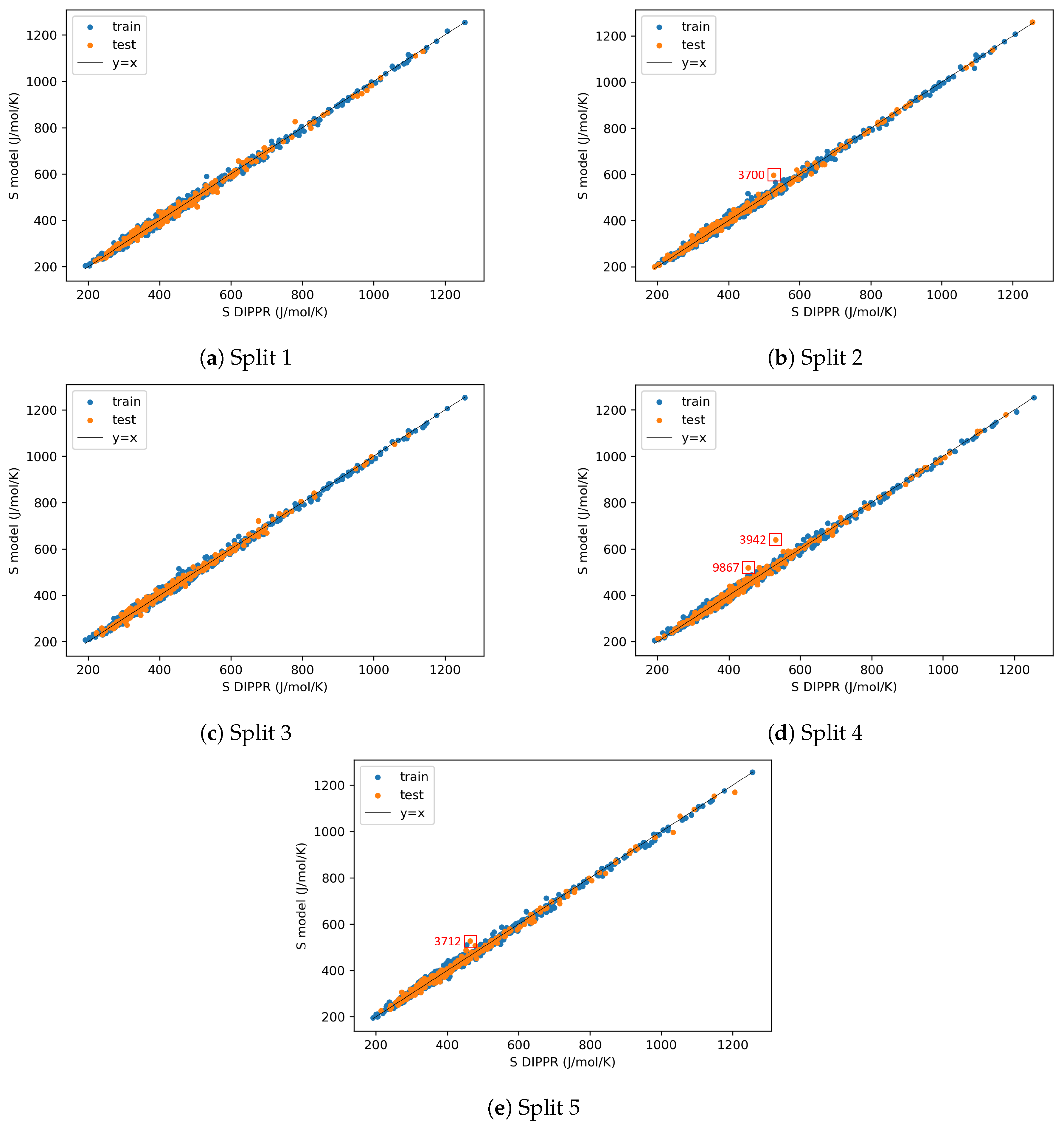
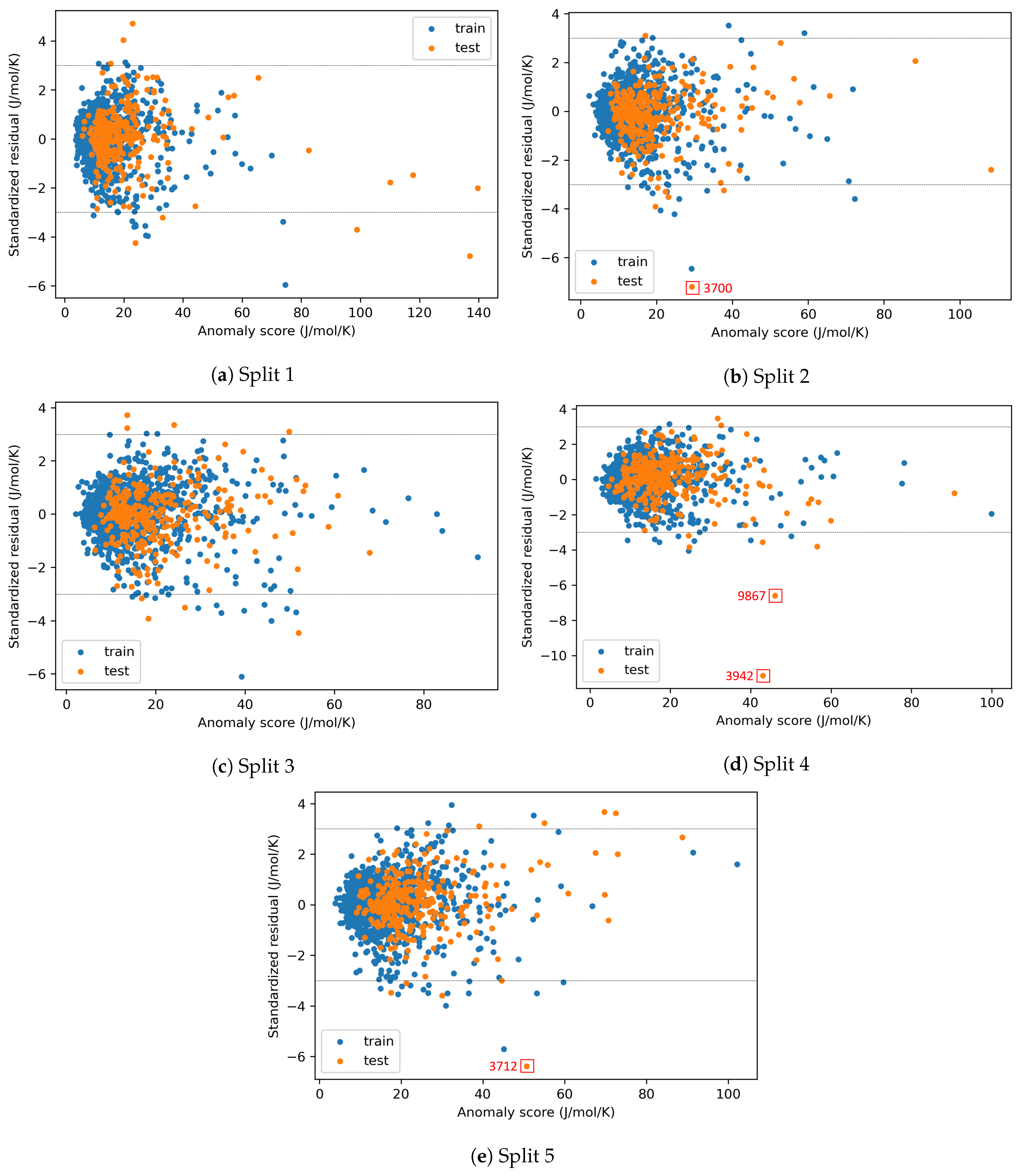
3.5. AD Definition during ML Model Deployment (Substudy 3)
4. Results
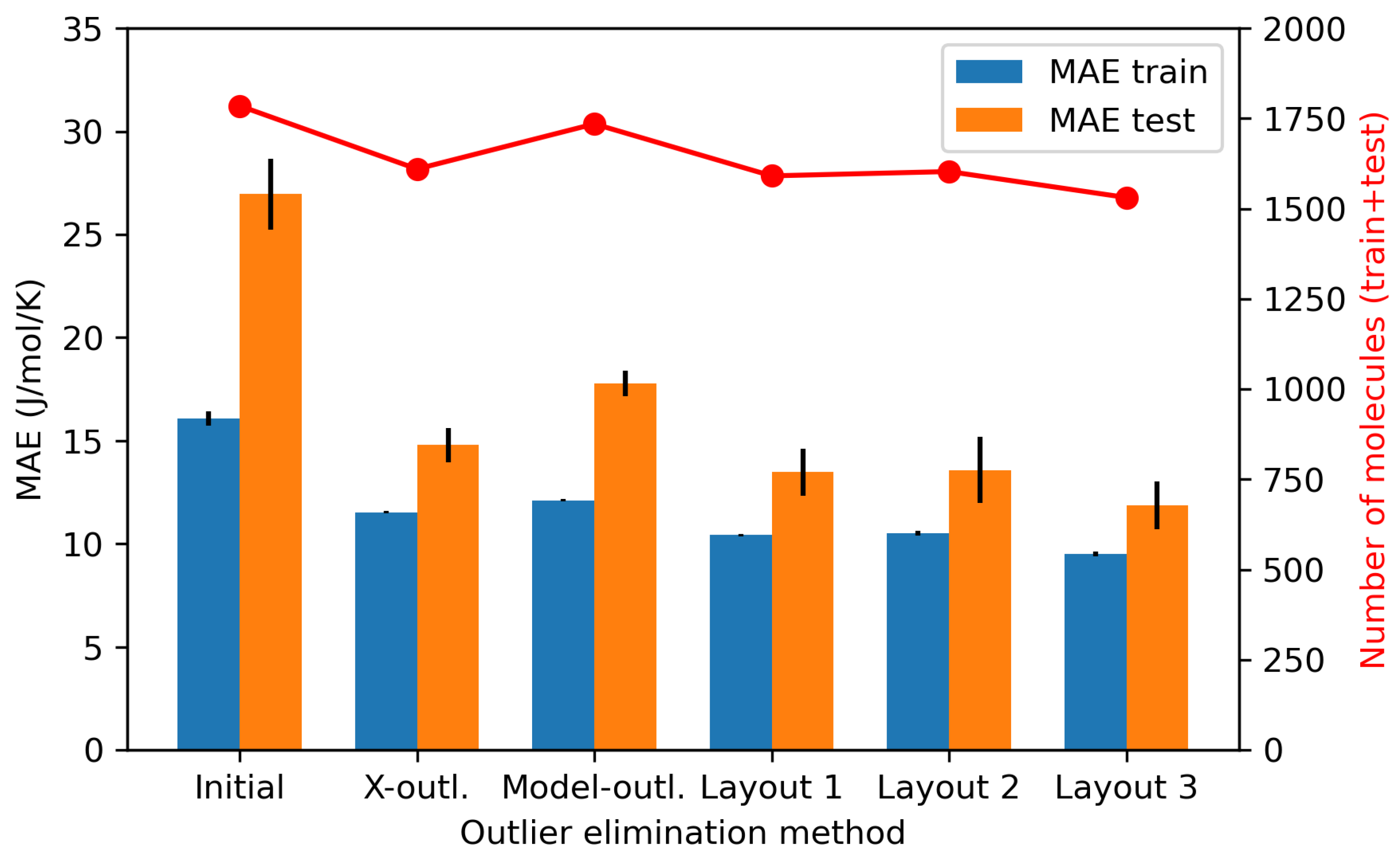
4.1. AD Definition as a Data Preprocessing Method (Substudy 1)
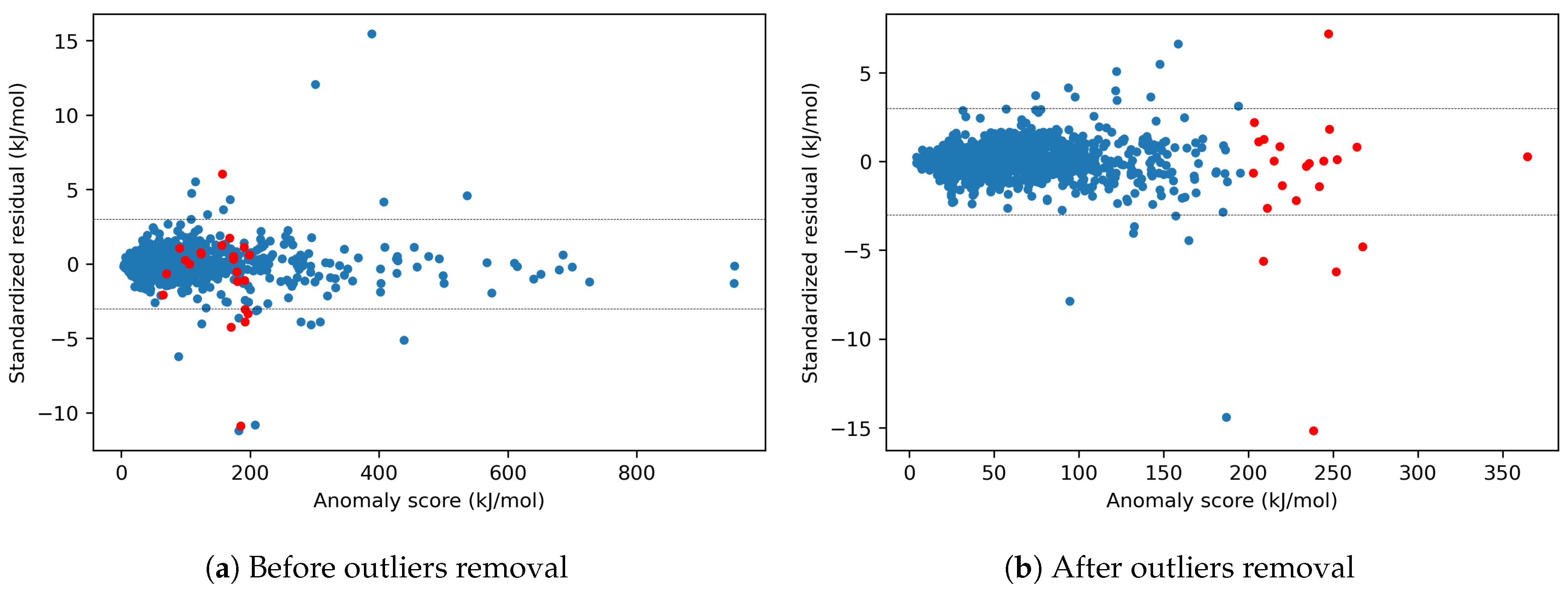
4.1.1. Outlier Detection
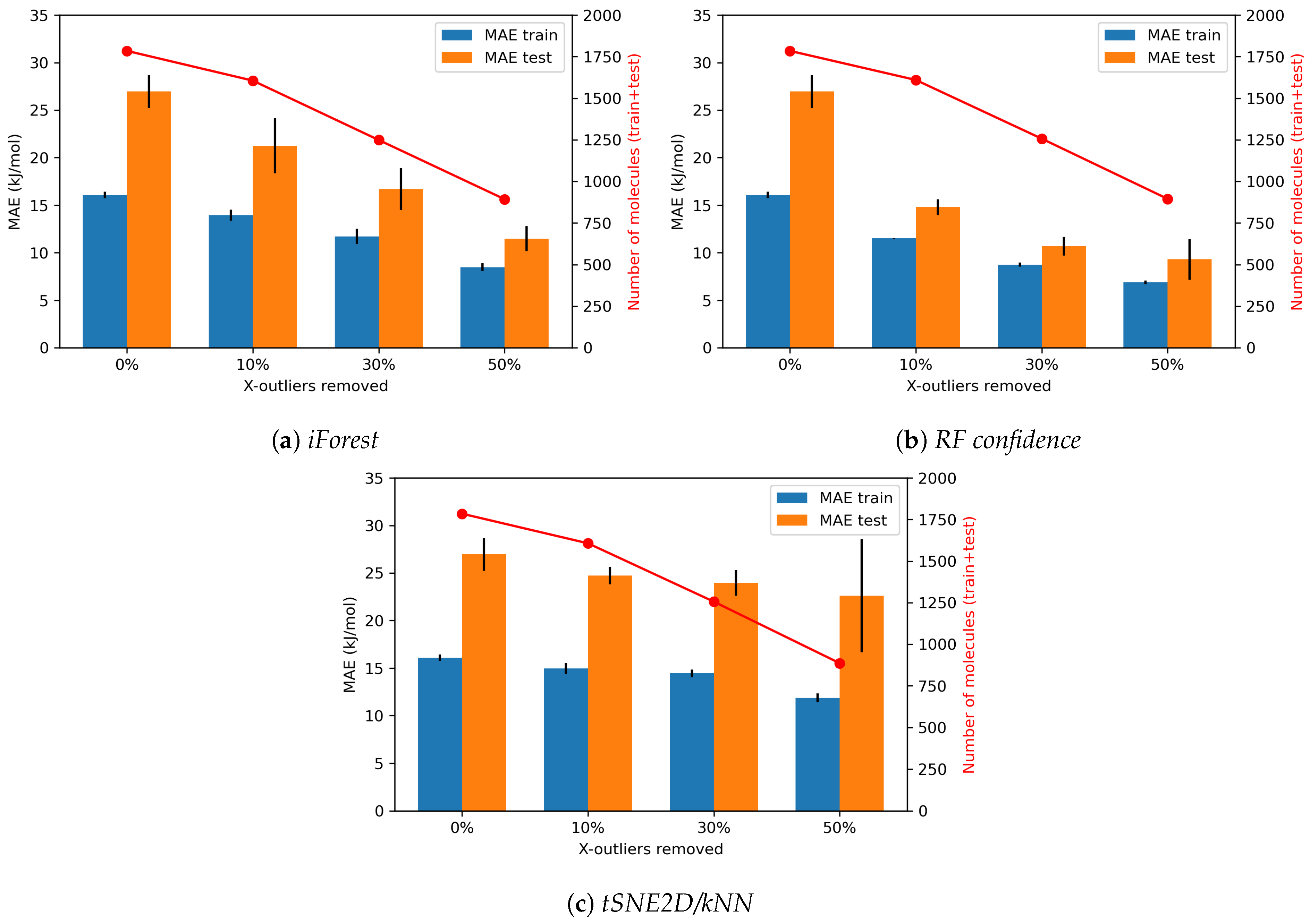
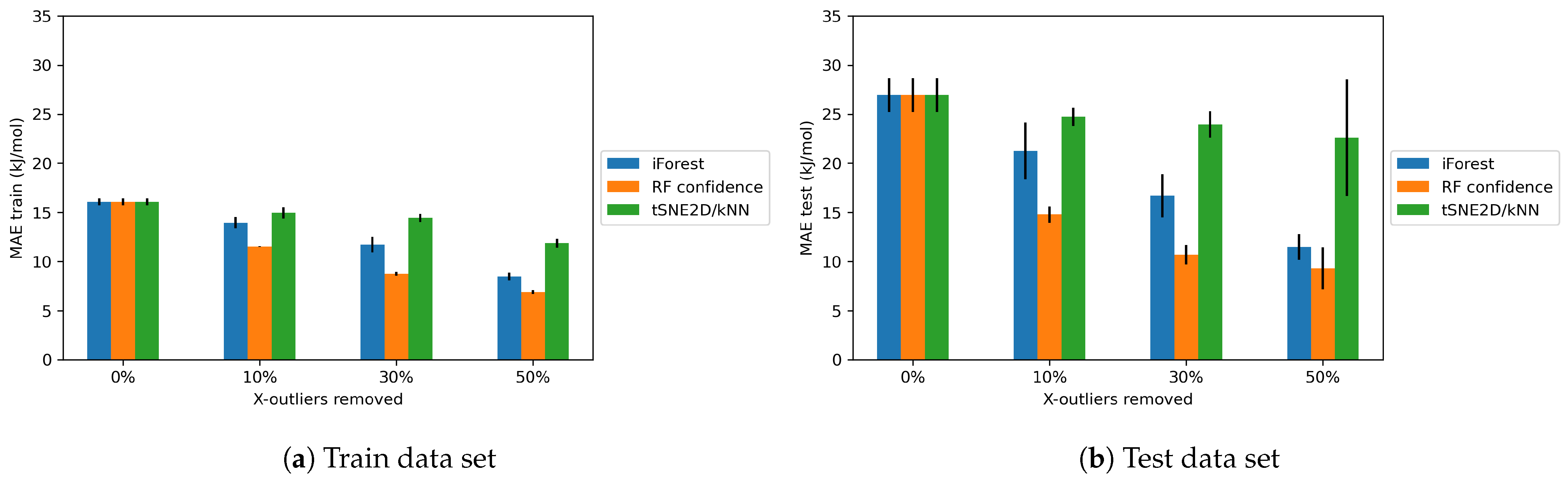
4.1.2. Effects of X-outliers on the Model Performance
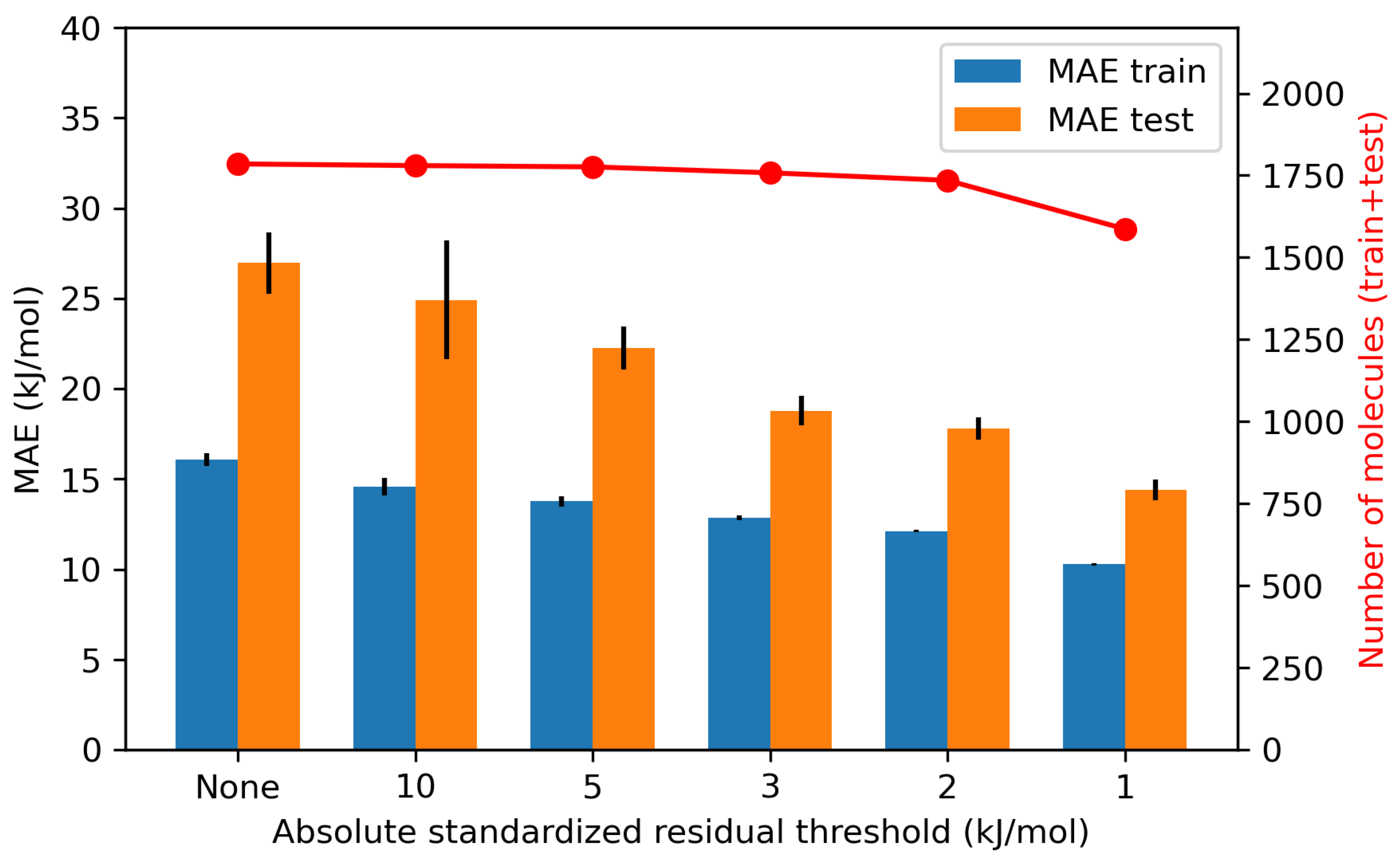
4.1.3. Effects of Model-Outliers on the Model Performance
4.1.4. Effects of X-outliers and Model-outliers on the Model Performance
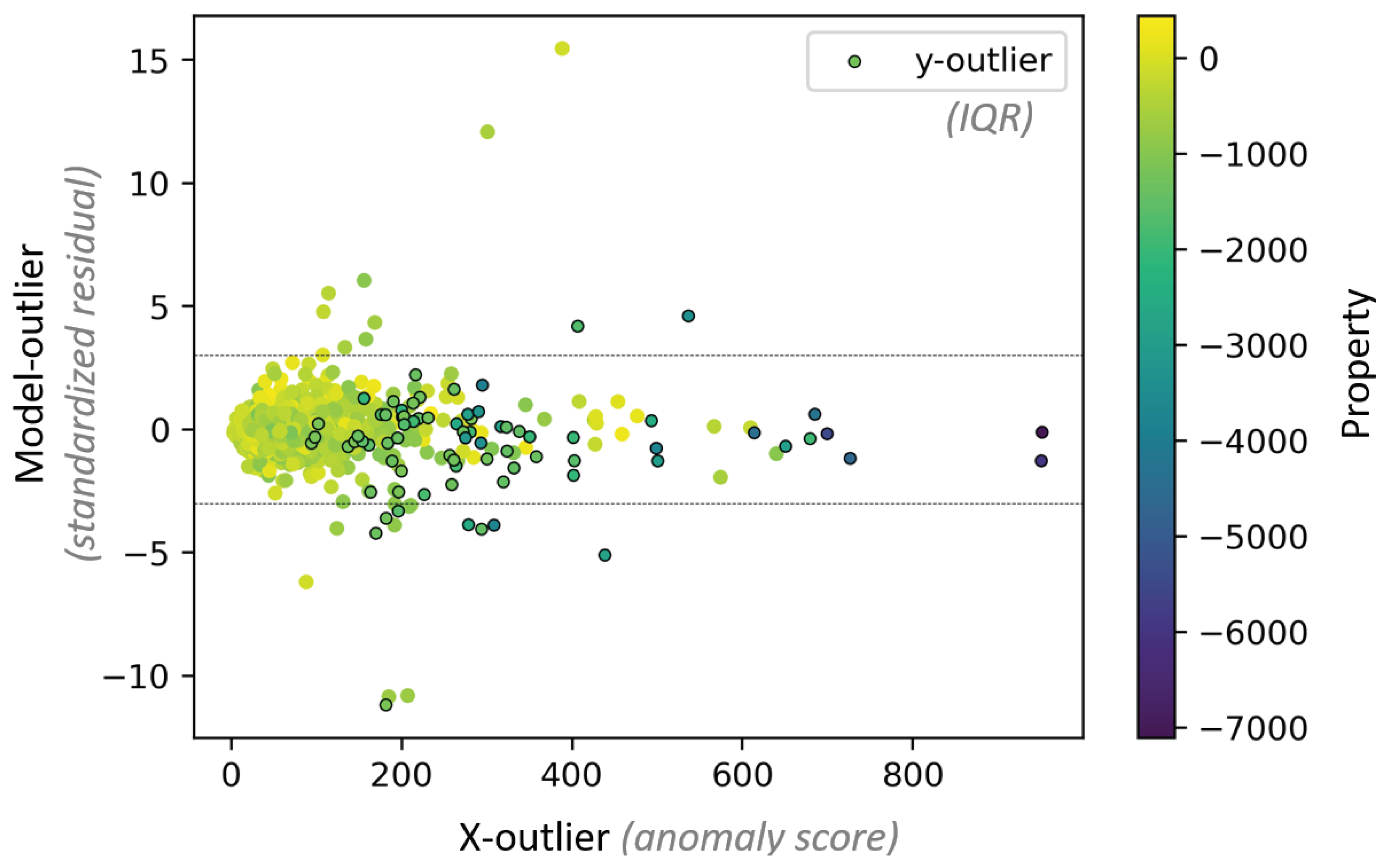
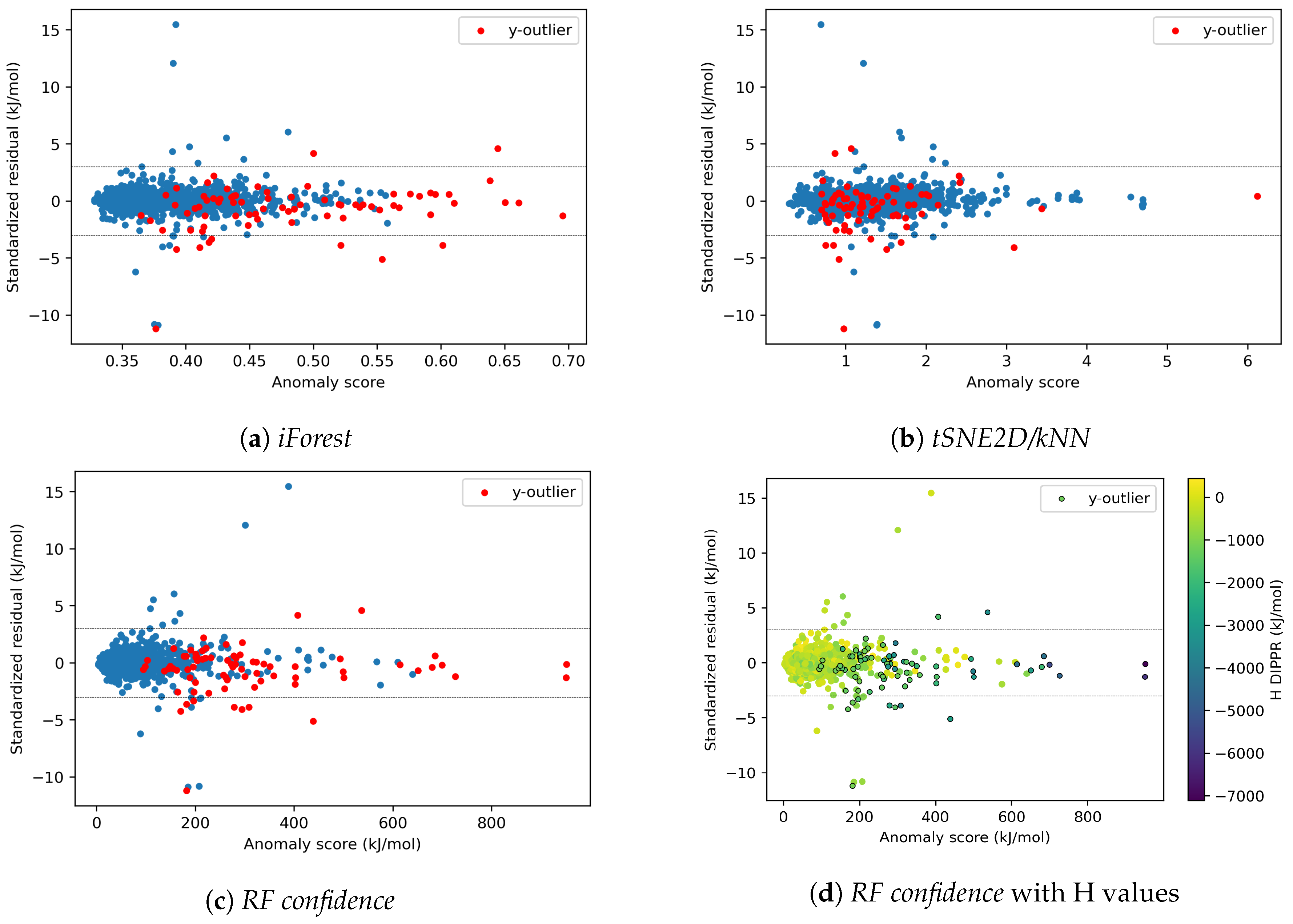
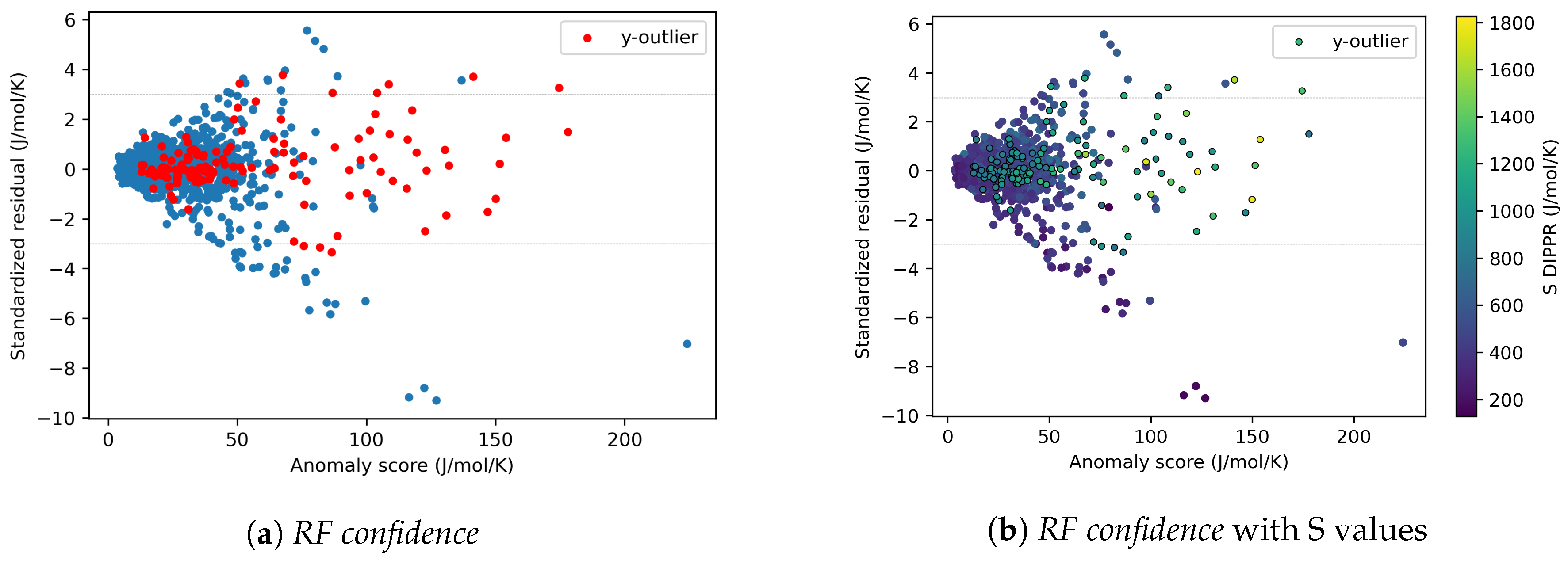
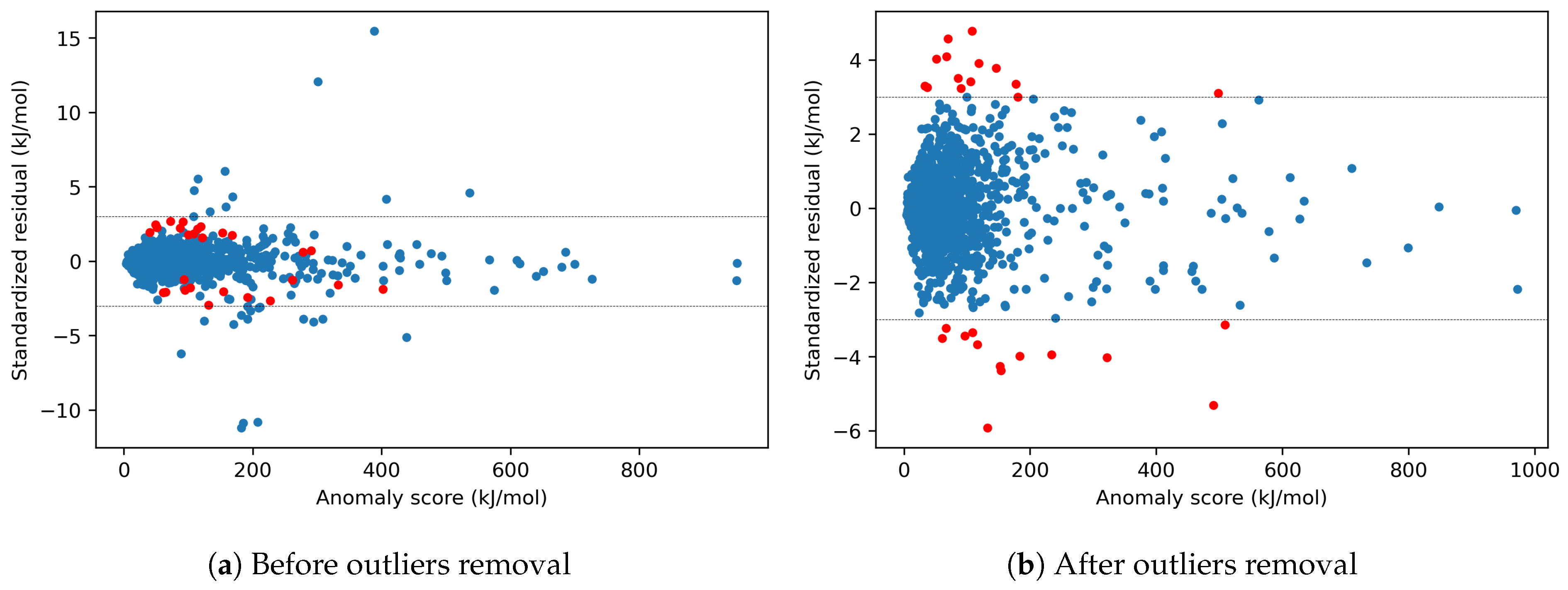
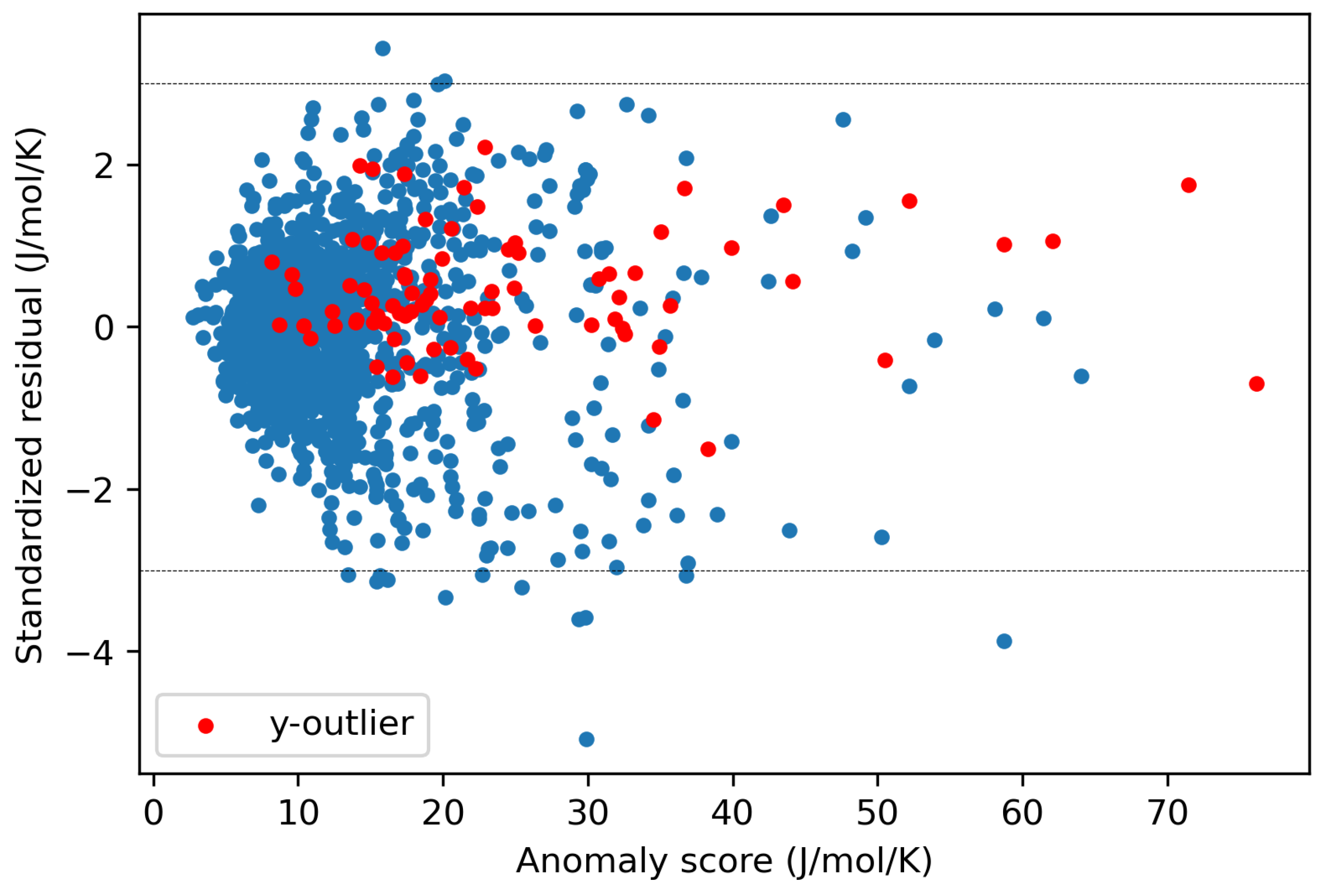
4.1.5. Effects of y-outliers on the Model Performance
4.2. AD Definition during ML Model Construction (Substudy 2)
4.2.1. Dimensionality Reduction on the Preprocessed Data without Outliers
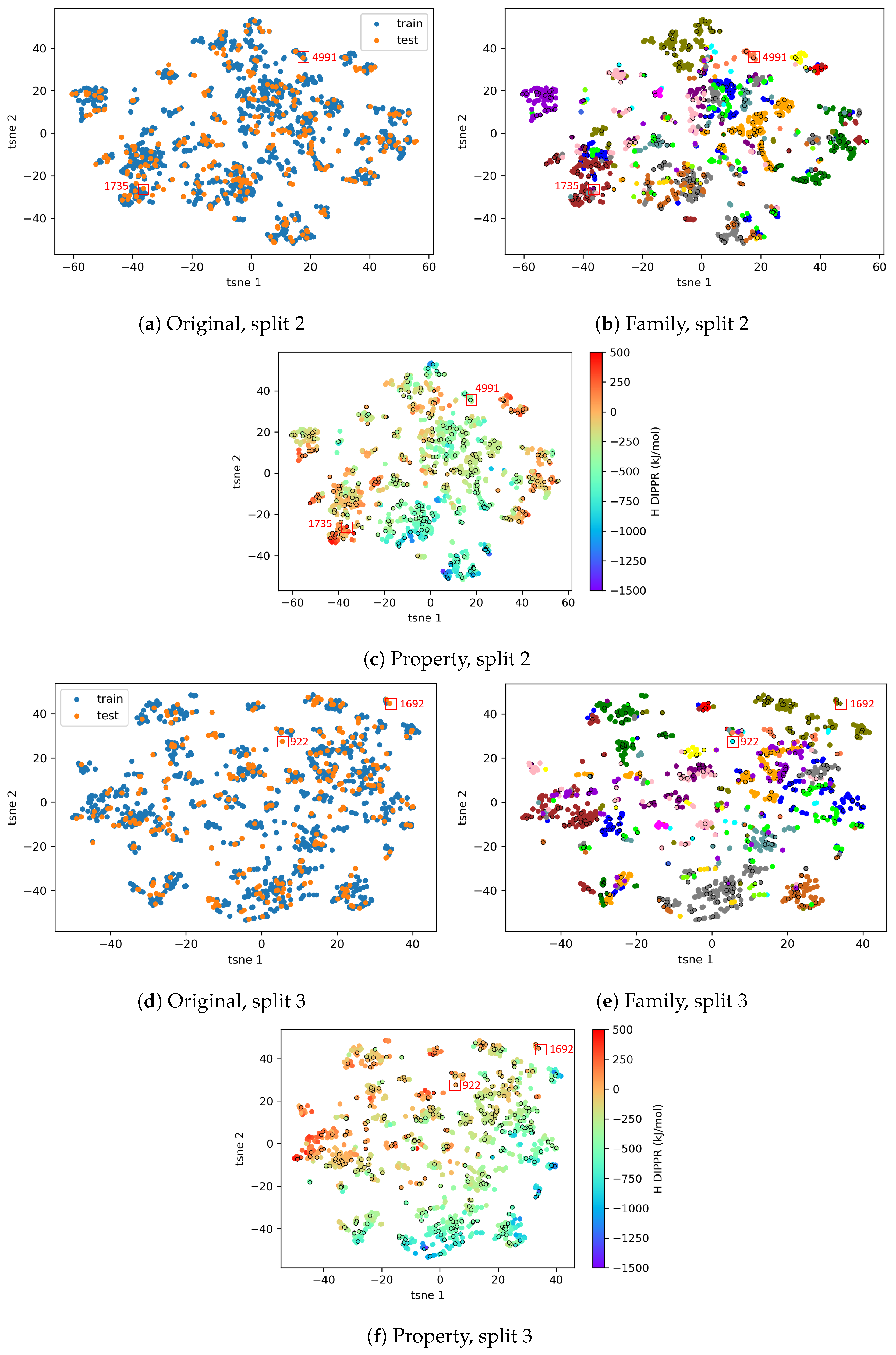
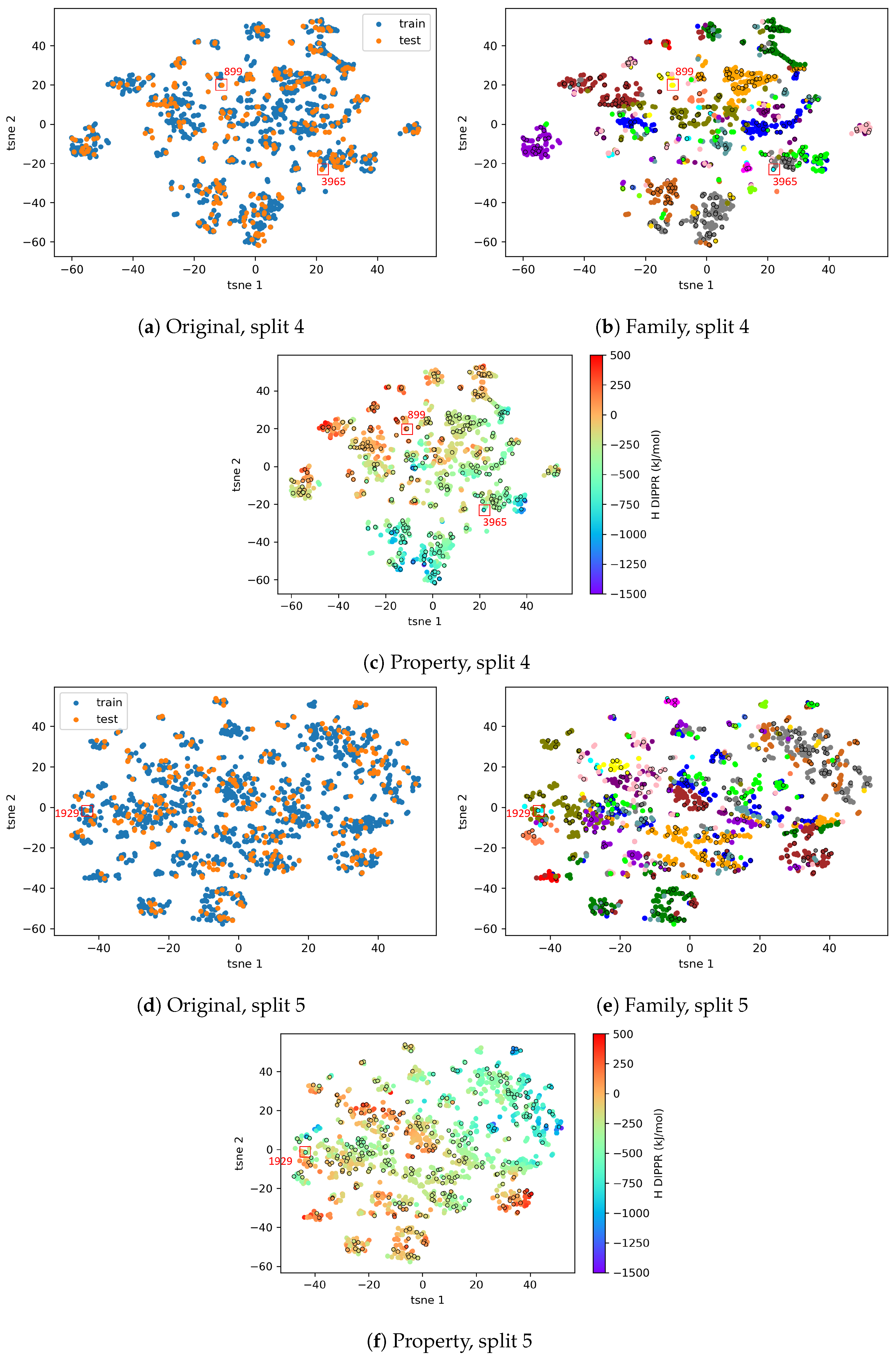
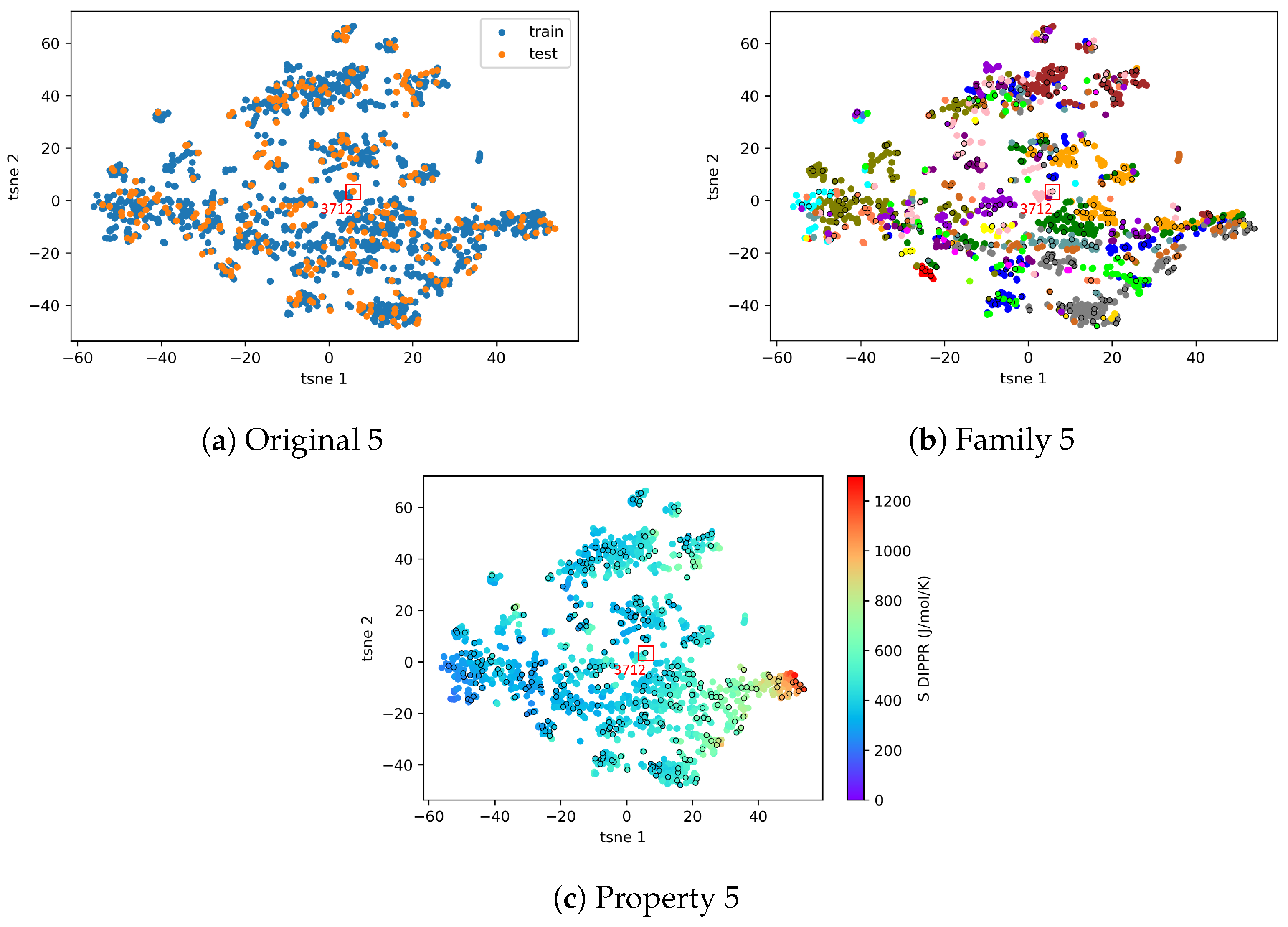
4.2.2. AD Visualization and Evaluation
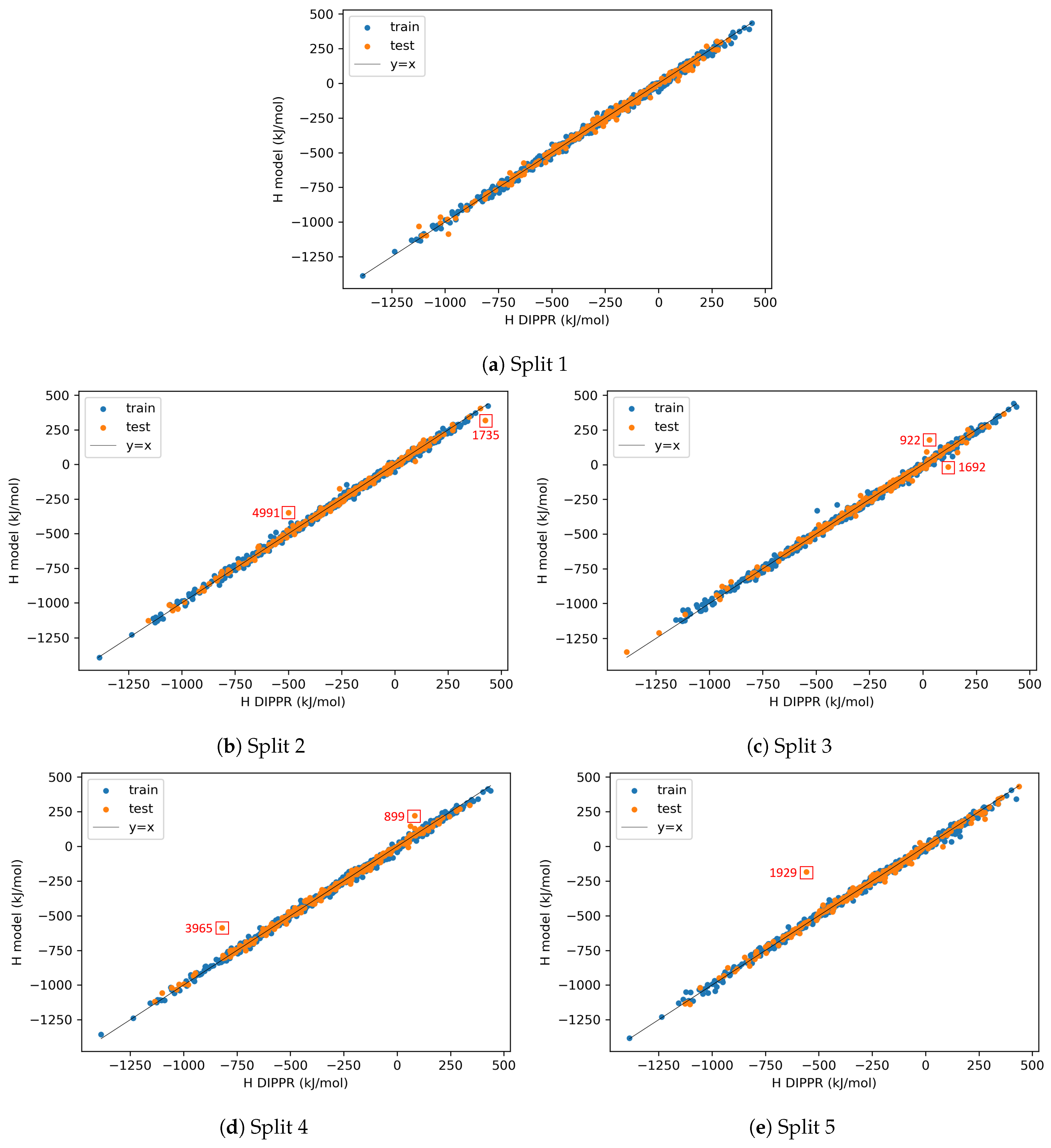
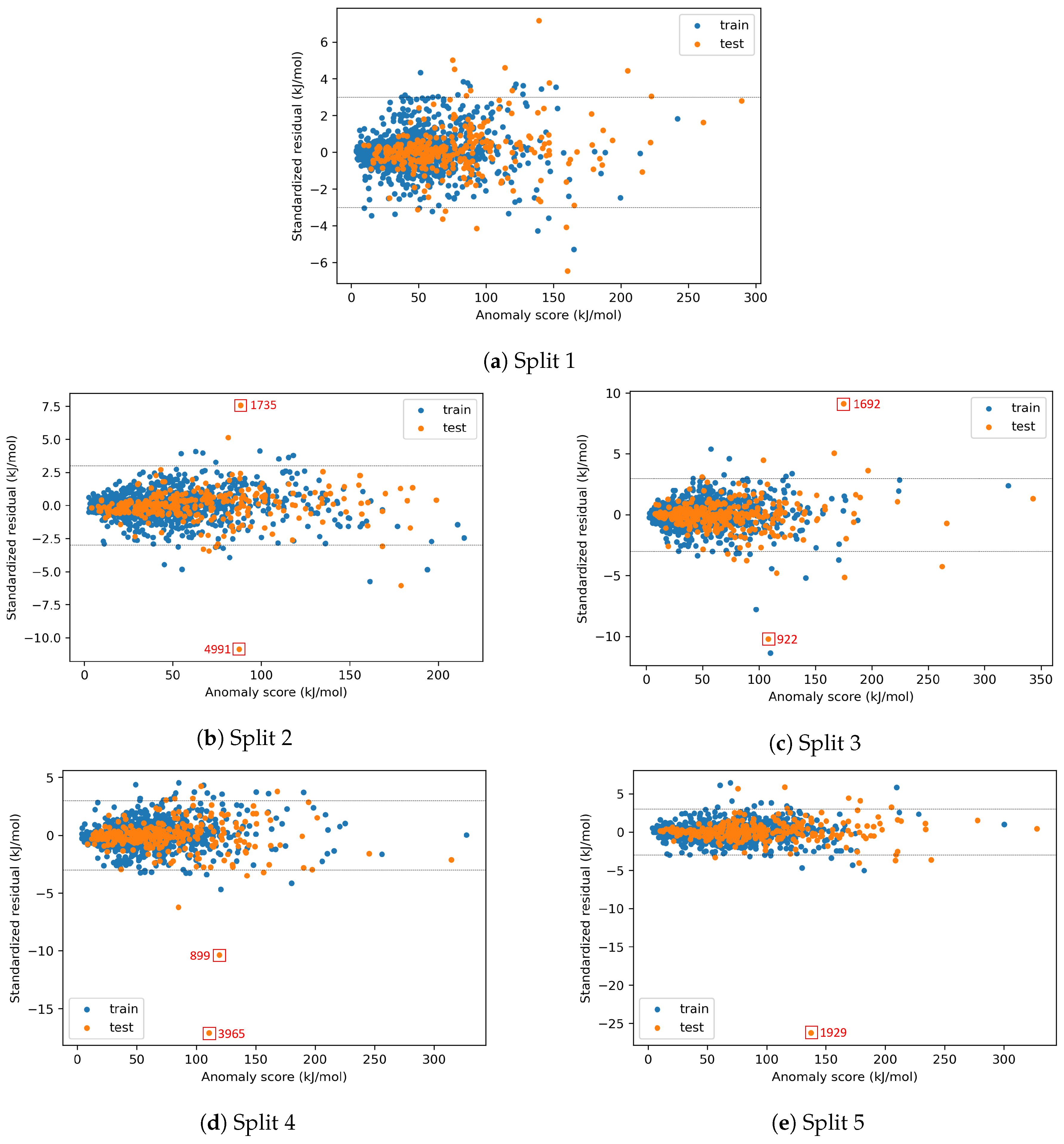
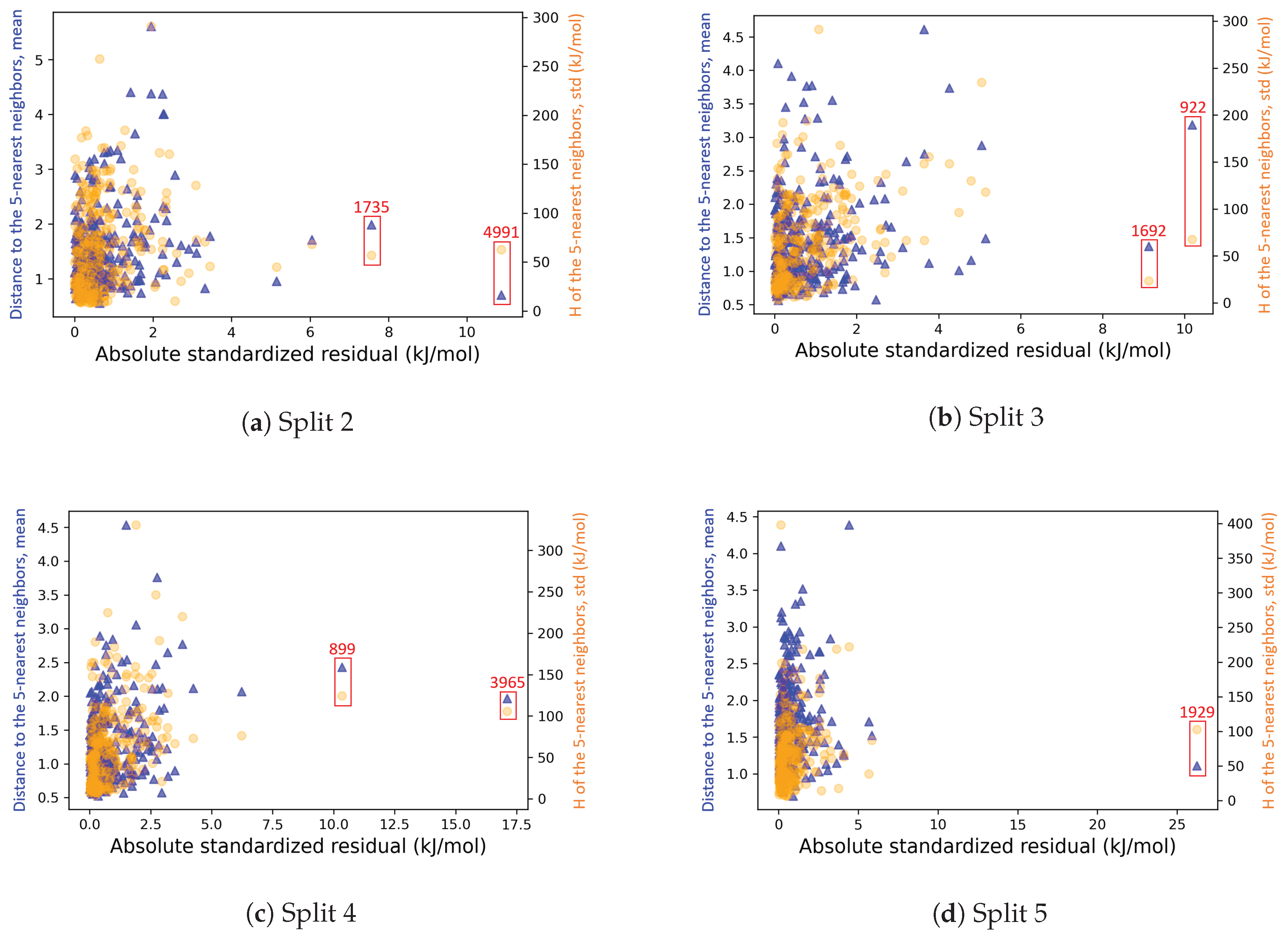
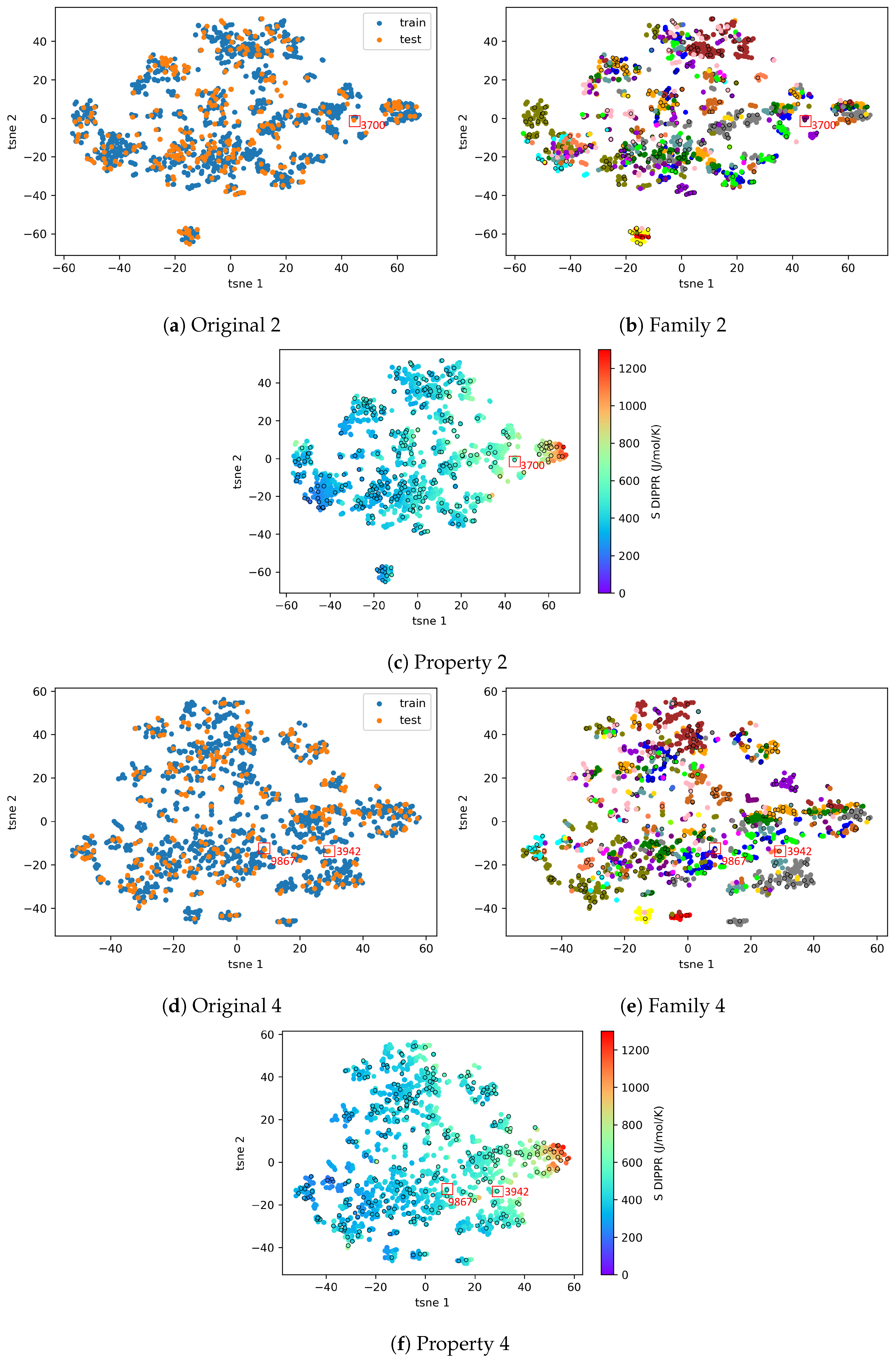
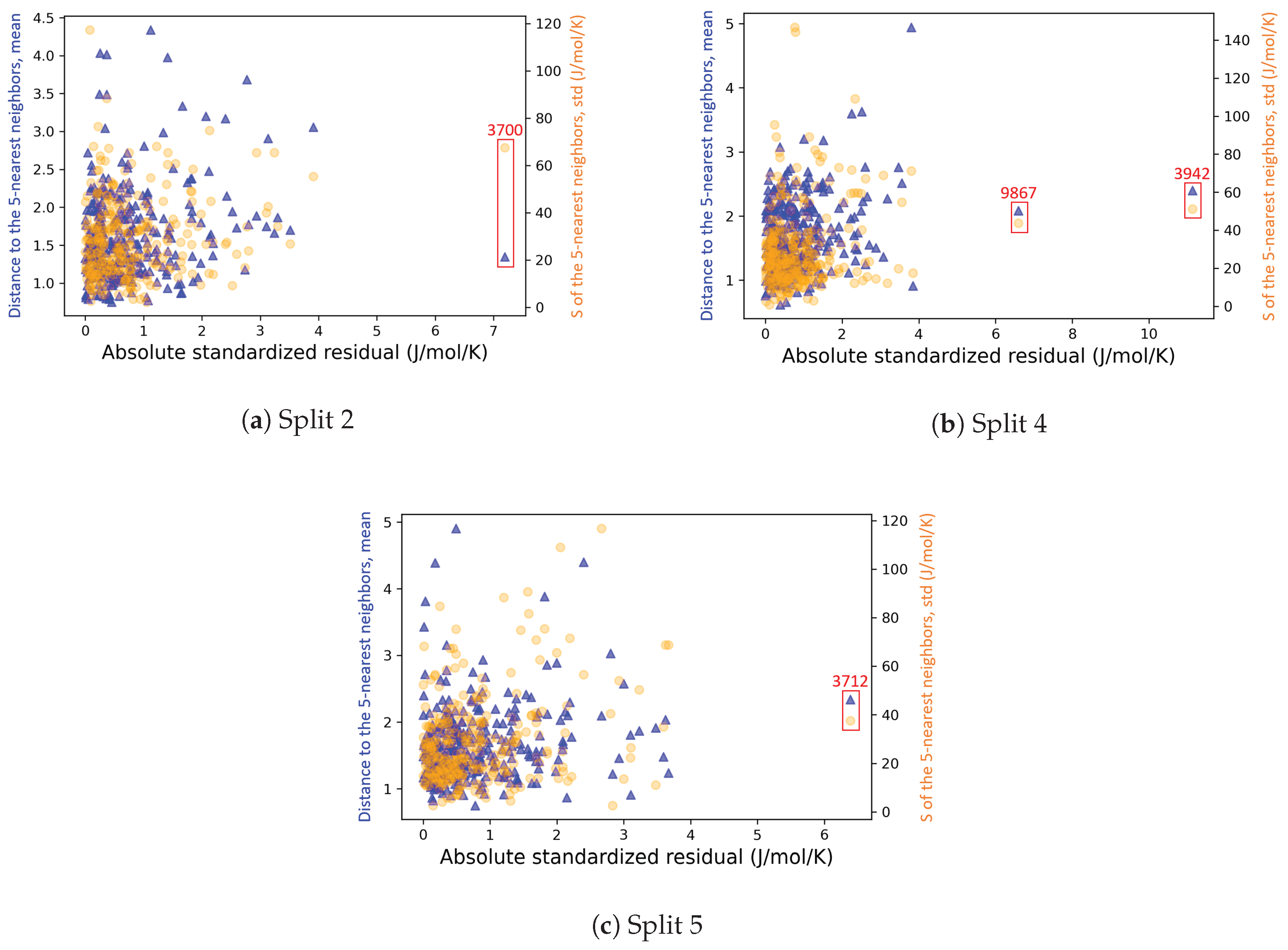
4.2.3. On the Understanding of the High Prediction Errors for Test Molecules
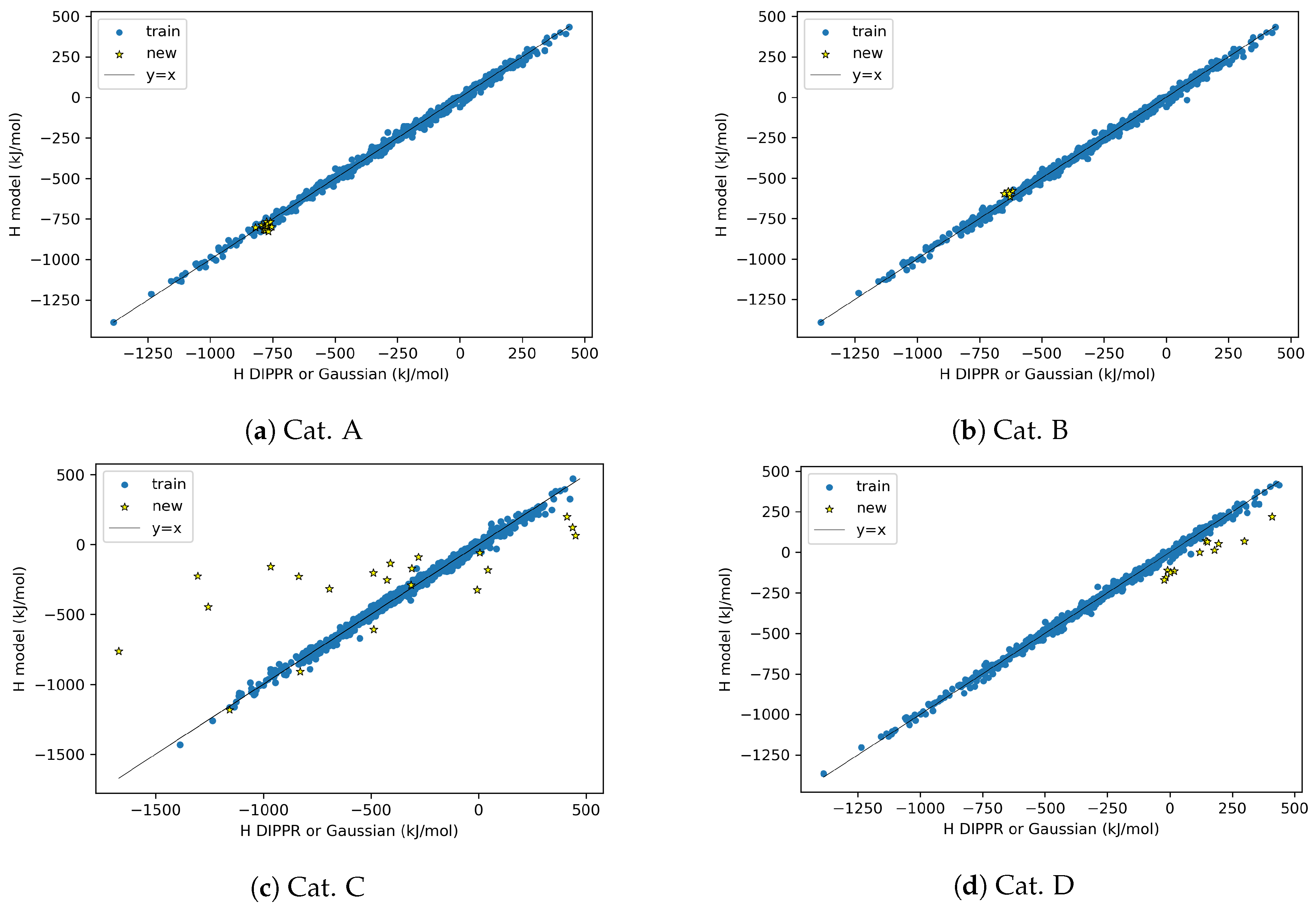
4.3. AD Definition during ML Model Deployment (Substudy 3)
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Applicability domain. |
| DIPPR | Design institute for physical properties. |
| DFT | Density functional theory. |
| H | Enthalpy for ideal gas at 298.15 K and 1 bar. |
| IQR | Interquartile range. |
| GA | Genetic algorithm. |
| GNN | Graph neural network. |
| GP | Gaussian processes. |
| iForest | Isolation forest. |
| kNN | k-nearest neighbors. |
| Lasso | Least absolute shrinkage and selection operator. |
| Mean absolute error. | |
| ML | Machine learning. |
| OECD | Organisation for economic co-operation and development. |
| PCs | Principal components. |
| PCA | Principal component analysis. |
| QSAR | Quantitative structure–activity relationship. |
| QSPR | Quantitative structure–property relationship. |
| Coefficient of determination. | |
| REACH | Registration, Evaluation, Authorization and Restriction of CHemicals. |
| RF | Random forest. |
| RF confidence | Random forest prediction confidence. |
| Root mean square error. | |
| S | Absolute entropy of ideal gas at 298.15 K and 1 bar. |
| tSNE | t-distributed stochastic neighbor embedding. |
Appendix A. Identification of Model-outliers and X-outliers in the Preprocessed Data for Enthalpy

| Absolute Standardized | Chemid | Family |
|---|---|---|
| Residual | ||
| 3608 | Halogen Compounds | |
| 2628 | Halogen Compounds | |
| 3862 | Polyfunctional Compounds | |
| 7886 | Polyfunctional Compounds | |
| 7887 | Polyfunctional Compounds | |
| 1840 | Sulfur Compounds | |
| 2254 | Organic Acids | |
| 1950 | Inorganic Compounds | |
| 1969 | Silicon Compounds | |
| 2283 | Organic Acids | |
| 3931 | Silicon Compounds | |
| 3948 | Silicon Compounds | |
| 3929 | Silicon Compounds | |
| 4994 | Silicon Compounds | |
| 9858 | Organic Salts | |
| 2619 | Halogen Compounds | |
| 6851 | Other Compounds | |
| 2995 | Silicon Compounds | |
| 3991 | Silicon Compounds | |
| 3958 | Silicon Compounds | |
| 2370 | Esters/Ethers | |
| 2653 | Halogen Compounds | |
| 2877 | Polyfunctional Compounds | |
| 3974 | Silicon Compounds | |
| 3898 | Inorganic Compounds | |
| 6850 | Organic Compounds | |
| 9866 | Nitrogen Compounds |
| N° | iForest | RF Confidence | tSNE2D/kNN | |||
|---|---|---|---|---|---|---|
| Chemid | Family | Chemid | Family | Chemid | Family | |
| 1 | 3933 | Silicon Compounds | 2624 | Halogen Compounds | 3977 | Silicon Compounds |
| 2 | 3932 | Silicon Compounds | 3933 | Silicon Compounds | 1509 | Halogen Compounds |
| 3 | 2624 | Halogen Compounds | 1627 | Halogen Compounds | 1866 | Halogen Compounds |
| 4 | 3931 | Silicon Compounds | 1631 | Halogen Compounds | 9879 | Nitrogen Compounds |
| 5 | 2991 | Silicon Compounds | 1626 | Halogen Compounds | 9877 | Nitrogen Compounds |
| 6 | 1631 | Halogen Compounds | 3881 | Polyfunctional Compounds | 90 | Alkanes |
| 7 | 3877 | Other Compounds | 1930 | Inorganic Compounds | 5878 | Organic Acids |
| 8 | 2995 | Silicon Compounds | 1864 | Halogen Compounds | 1097 | Ketones/Aldehydes |
| 9 | 3348 | Esters/Ethers | 3932 | Silicon Compounds | 3056 | Ketones/Aldehydes |
| 10 | 1627 | Halogen Compounds | 834 | Alkanes | 7883 | Nitrogen Compounds |
Appendix B. Most Represented Families in the Eliminated Outliers (Layout 3) for Enthalpy and Entropy
| Enthalpy | Entropy | ||
|---|---|---|---|
| Halogen Compounds | 24% | Esters/Ethers | 20% |
| Silicon Compounds | 16% | Silicon Compounds | 13% |
| Esters/Ethers | 10% | Polyfunctional Compounds | 9% |
| Nitrogen Compounds | 9% | Nitrogen Compounds | 9% |
| Inorganic Compounds | 7% | Aromatics | 7% |
| Total | 66% | Total | 58% |
Appendix C. AD Definition for Entropy During Model Construction





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
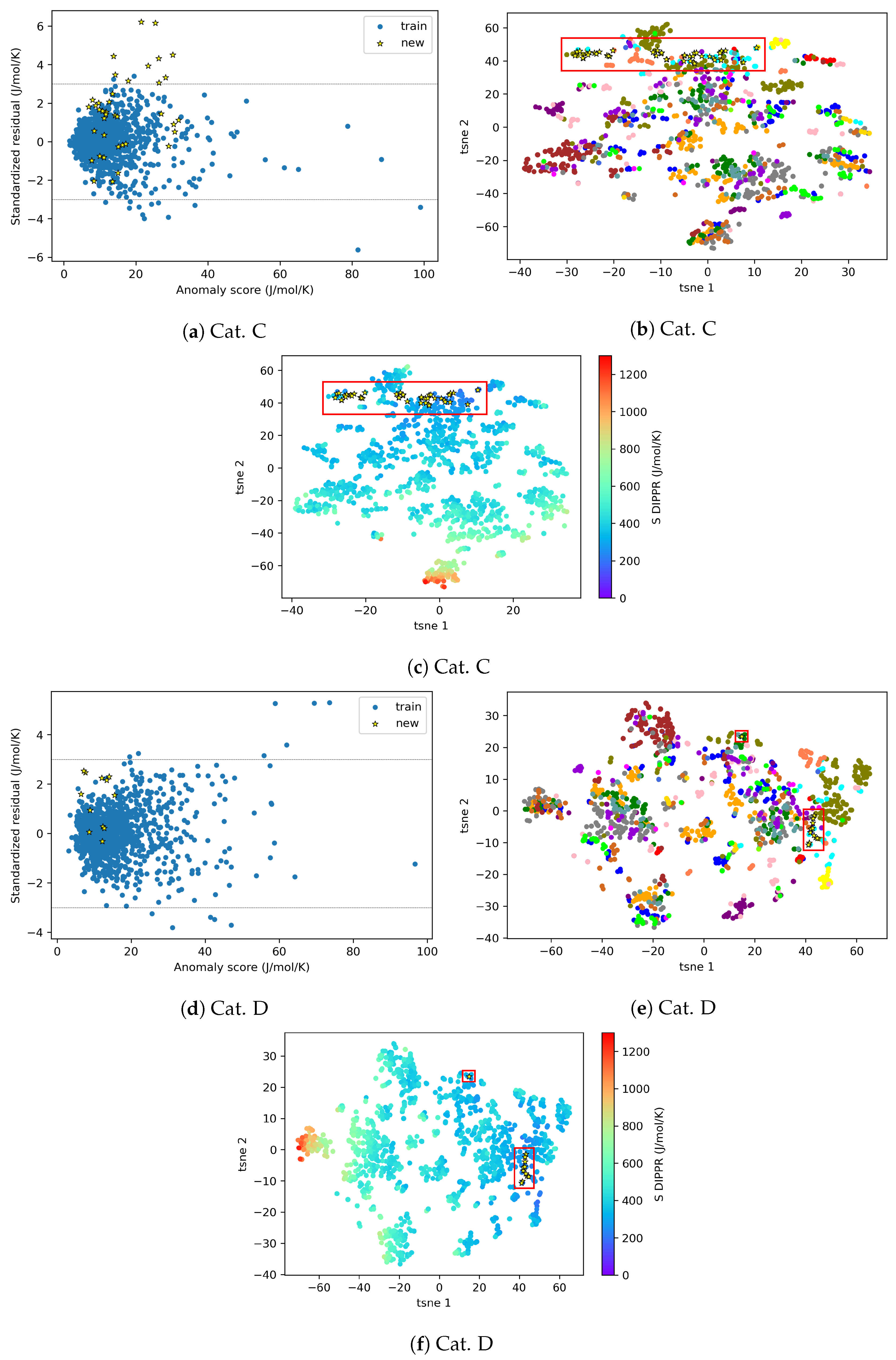{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

References
- Netzeva, T.I.; Worth, A.P.; Aldenberg, T.; Benigni, R.; Cronin, M.T.; Gramatica, P.; Jaworska, J.S.; Kahn, S.; Klopman, G.; Marchant, C.A.; et al. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. ATLA Altern. Lab. Anim. 2005, 33, 155–173. [Google Scholar] [CrossRef]
- McCartney, M.; Haeringer, M.; Polifke, W. Comparison of Machine Learning Algorithms in the Interpolation and Extrapolation of Flame Describing Functions. J. Eng. Gas Turbines Power 2020, 142, 061009. [Google Scholar] [CrossRef]
- Cao, X.; Yousefzadeh, R. Extrapolation and AI transparency: Why machine learning models should reveal when they make decisions beyond their training. Big Data Soc. 2023, 10, 20539517231169731. [Google Scholar] [CrossRef]
- European Commission Environment Directorate General. Guidance Document on the Validation of (Quantitative)Structure-Activity Relationships [(Q)Sar] Models; OECD: Paris, France, 2014; pp. 1–154. [Google Scholar]
- Dearden, J.C.; Cronin, M.T.; Kaiser, K.L. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR). SAR QSA Environ. Res. 2009, 20, 241–266. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Smith, I.F.C. Extrapolation with machine learning based early-stage energy prediction models. In Proceedings of the 2023 European Conference on Computing in Construction and the 40th International CIB W78 Conference, Crete, Greece, 10–12 July 2023; Volume 4. [Google Scholar] [CrossRef]
- Muckley, E.S.; Saal, J.E.; Meredig, B.; Roper, C.S.; Martin, J.H. Interpretable models for extrapolation in scientific machine learning. Digit. Discov. 2023, 2, 1425–1435. [Google Scholar] [CrossRef]
- Hoaglin, D.C.; Kempthorne, P.J. Influential Observations, High Leverage Points, and Outliers in Linear Regression: Comment. Stat. Sci. 1986, 1, 408–412. [Google Scholar] [CrossRef]
- Aggarwal, C.C.; Yu, P.S. Outlier detection for high dimensional data. In Proceedings of the ACM SIGMOD International Conference on Management of Data, Santa Barbara, CA, USA, 21–24 May 2001; pp. 37–46. [Google Scholar] [CrossRef]
- Akoglu, L.; Tong, H.; Koutra, D. Graph based anomaly detection and description: A survey. Data Min. Knowl. Discov. 2015, 29, 626–688. [Google Scholar] [CrossRef]
- Souiden, I.; Omri, M.N.; Brahmi, Z. A survey of outlier detection in high dimensional data streams. Comput. Sci. Rev. 2022, 44, 100463. [Google Scholar] [CrossRef]
- Smiti, A. A critical overview of outlier detection methods. Comput. Sci. Rev. 2020, 38, 100306. [Google Scholar] [CrossRef]
- Cao, D.S.; Liang, Y.Z.; Xu, Q.S.; Li, H.D.; Chen, X. A New Strategy of Outlier Detection for QSAR/QSPR. J. Comput. Chem. 2010, 31, 592–602. [Google Scholar] [CrossRef]
- De Maesschalck, R.; Estienne, F.; Verdu-Andres, J.; Candolfi, A.; Centner, V.; Despagne, F.; Jouan-Rimbaud, D.; Walczak, B.; Massart, D.L.; De Jong, S.; et al. The development of calibration models for spectroscopic data using principal component regression [Review]. Internet J. Chem. 1999, 2, 1–21. [Google Scholar]
- Trinh, C.; Tbatou, Y.; Lasala, S.; Herbinet, O.; Meimaroglou, D. On the Development of Descriptor-Based Machine Learning Models for Thermodynamic Properties. Part 1—From Data Collection to Model Construction: Understanding of the Methods and their Effects. Processes 2023, 11, 3325. [Google Scholar] [CrossRef]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. ATLA Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Mathea, M.; Klingspohn, W.; Baumann, K. Chemoinformatic Classification Methods and their Applicability Domain. Mol. Inform. 2016, 35, 160–180. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Yalamanchi, K.K.; van Oudenhoven, V.C.; Tutino, F.; Monge-Palacios, M.; Alshehri, A.; Gao, X.; Sarathy, S.M. Machine Learning to Predict Standard Enthalpy of Formation of Hydrocarbons. J. Phys. Chem. A 2019, 123, 8305–8313. [Google Scholar] [CrossRef]
- Yalamanchi, K.K.; Monge-Palacios, M.; van Oudenhoven, V.C.; Gao, X.; Sarathy, S.M. Data Science Approach to Estimate Enthalpy of Formation of Cyclic Hydrocarbons. J. Phys. Chem. A 2020, 124, 6270–6276. [Google Scholar] [CrossRef]
- Aldosari, M.N.; Yalamanchi, K.K.; Gao, X.; Sarathy, S.M. Predicting entropy and heat capacity of hydrocarbons using machine learning. Energy AI 2021, 4, 100054. [Google Scholar] [CrossRef]
- Aouichaoui, A.R.; Fan, F.; Abildskov, J.; Sin, G. Application of interpretable group-embedded graph neural networks for pure compound properties. Comput. Chem. Eng. 2023, 176, 108291. [Google Scholar] [CrossRef]
- Balestriero, R.; Pesenti, J.; LeCun, Y. Learning in High Dimension Always Amounts to Extrapolation. arXiv 2021, arXiv:2110.09485. [Google Scholar]
- Ghorbani, H. Mahalanobis Distance and Its Application for detecting multivariate outliers. Ser. Math. Inform. 2019, 34, 583–595. [Google Scholar] [CrossRef]
- De Maesschalck, R.; Jouan-Rimbaud, D.; Massart, D.L. The Mahalanobis distance. Chemom. Intell. Lab. Syst. 2000, 50, 1–18. [Google Scholar] [CrossRef]
- Aouichaoui, A.R.; Fan, F.; Mansouri, S.S.; Abildskov, J.; Sin, G. Combining Group-Contribution Concept and Graph Neural Networks Toward Interpretable Molecular Property Models. J. Chem. Inf. Model. 2023, 63, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Bertola, M. Alvascience: A New Software Suite for the QSAR Workflow Applied to the Blood–Brain Barrier Permeability. Int. J. Mol. Sci. 2022, 23, 12882. [Google Scholar] [CrossRef] [PubMed]
- Huoyu, R.; Zhiqiang, Z.; Zhanggao, L.; Zhenzhen, X. Quantitative structure–property relationship for the critical temperature of saturated monobasic ketones, aldehydes, and ethers with molecular descriptors. Int. J. Quantum Chem. 2022, 122, 1–10. [Google Scholar] [CrossRef]
- Cao, L.; Zhu, P.; Zhao, Y.; Zhao, J. Using machine learning and quantum chemistry descriptors to predict the toxicity of ionic liquids. J. Hazard. Mater. 2018, 352, 17–26. [Google Scholar] [CrossRef]
- Yousefinejad, S.; Mahboubifar, M.; Eskandari, R. Quantitative structure-activity relationship to predict the anti-malarial activity in a set of new imidazolopiperazines based on artificial neural networks. Malar. J. 2019, 18, 1–17. [Google Scholar] [CrossRef]
- Asadollahi, T.; Dadfarnia, S.; Shabani, A.M.H.; Ghasemi, J.B.; Sarkhosh, M. QSAR models for cxcr2 receptor antagonists based on the genetic algorithm for data preprocessing prior to application of the pls linear regression method and design of the new compounds using in silico virtual screening. Molecules 2011, 16, 1928–1955. [Google Scholar] [CrossRef]
- Kim, M.G. Sources of High Leverage in Linear Regression Model. J. Appl. Math. Inform. 2004, 16, 509–513. [Google Scholar]
- Leys, C.; Klein, O.; Dominicy, Y.; Ley, C. Detecting multivariate outliers: Use a robust variant of the Mahalanobis distance. J. Exp. Soc. Psychol. 2018, 74, 150–156. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Varamesh, A.; Hemmati-Sarapardeh, A.; Dabir, B.; Mohammadi, A.H. Development of robust generalized models for estimating the normal boiling points of pure chemical compounds. J. Mol. Liq. 2017, 242, 59–69. [Google Scholar] [CrossRef]
- Sabando, M.V.; Ponzoni, I.; Soto, A.J. Neural-based approaches to overcome feature selection and applicability domain in drug-related property prediction. Appl. Soft Comput. J. 2019, 85, 105777. [Google Scholar] [CrossRef]
- Huang, J.; Fan, X. Reliably assessing prediction reliability for high dimensional QSAR data. Mol. Divers. 2013, 17, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Rakhimbekova, A.; Madzhidov, T.; Nugmanov, R.I.; Baskin, I.; Varnek, A.; Rakhimbekova, A.; Madzhidov, T.; Nugmanov, R.I.; Gimadiev, T.; Baskin, I. Comprehensive Analysis of Applicability Domains of QSPR Models for Chemical Reactions. Int. J. Mol. Sci. 2021, 21, 5542. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Funatsu, K. Applicability domain based on ensemble learning in classification and regression analyses. J. Chem. Inf. Model. 2014, 54, 2469–2482. [Google Scholar] [CrossRef]
- Rasmussen, C.E.; Williams, C.K.I. Gaussian Processes for Machine Learning; MIT Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Sushko, I. Applicability Domain of QSAR Models. Ph.D. Thesis, Technical University of Munich, Munich, Germany, 2011. [Google Scholar]
- Kamalov, F.; Leung, H.H. Outlier Detection in High Dimensional Data. J. Inf. Knowl. Manag. 2020, 19, 1–15. [Google Scholar] [CrossRef]
- Riahi-Madvar, M.; Nasersharif, B.; Azirani, A.A. Subspace outlier detection in high dimensional data using ensemble of PCA-based subspaces. In Proceedings of the 26th International Computer Conference, Computer Society of Iran, CSICC 2021, Tehran, Iran, 3–4 March 2021. [Google Scholar] [CrossRef]
- Kriegel, H.P.; Kr, P.; Schubert, E.; Zimek, A. Outlier Detection in Axis-Parallel Subspaces of High Dimensional Data; Springer: Berlin Heidelberg, Germany, 2009; Volume 1, pp. 831–838. [Google Scholar]
- Filzmoser, P.; Maronna, R.; Werner, M. Outlier identification in high dimensions. Comput. Stat. Data Anal. 2008, 52, 1694–1711. [Google Scholar] [CrossRef]
- Angiulli, F.; Pizzuti, C. Fast outlier detection in high dimensional spaces. In Proceedings of the Principles of Data Mining and Knowledge Discovery, 6th European Conference PKDD, Helsinki, Finland, 19–23 August 2002; pp. 29–41. [Google Scholar]
- Kriegel, H.P.; Schubert, M.; Zimek, A. Angle-based outlier detection in high-dimensional data. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Las Vegas, NV, USA, 24–27 August 2008; pp. 444–452. [Google Scholar] [CrossRef]
- Liu, F.T.; Ting, K.M.; Zhou, Z.H. Isolation-based anomaly detection. ACM Trans. Knowl. Discov. Data 2012, 6, 1–39. [Google Scholar] [CrossRef]
- Thudumu, S.; Branch, P.; Jin, J.; Singh, J.J. A comprehensive survey of anomaly detection techniques for high dimensional big data. J. Big Data 2020, 7, 42. [Google Scholar] [CrossRef]
- Xu, X.; Liu, H.; Li, L.; Yao, M. A comparison of outlier detection techniques for high-dimensional data. Int. J. Comput. Intell. Syst. 2018, 11, 652–662. [Google Scholar] [CrossRef]
- Zimek, A.; Schubert, E.; Kriegel, H.P. A survey on unsupervised outlier detection in high-dimensional numerical data. Stat. Anal. Data Min. 2012, 5, 363–387. [Google Scholar] [CrossRef]
- Erfani, S.M.; Rajasegarar, S.; Karunasekera, S.; Leckie, C. High-dimensional and large-scale anomaly detection using a linear one-class SVM with deep learning. Pattern Recognit. 2016, 58, 121–134. [Google Scholar] [CrossRef]
- Alvascience, AlvaDesc (Software for Molecular Descriptors Calculation), Version 2.0.8. 2021. Available online: https://www.alvascience.com (accessed on 1 January 2023).
- Mauri, A. alvaDesc: A tool to calculate and analyze molecular descriptors and fingerprints. Methods Pharmacol. Toxicol. 2020, 2, 801–820. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Gaussian, Inc. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. [Google Scholar] [CrossRef]
- Becke, A.D. Thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Miyoshi, A. GPOP Software, Rev. 2022.01.20m1. Available online: http://akrmys.com/gpop/ (accessed on 1 January 2023).
- Non-Positive Definite Covariance Matrices. Available online: https://www.value-at-risk.net/non-positive-definite-covariance-matrices (accessed on 1 June 2023).
- Cruz-Monteagudo, M.; Medina-Franco, J.L.; Pérez-Castillo, Y.; Nicolotti, O.; Cordeiro, M.N.D.; Borges, F. Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde? Drug Discov. Today 2014, 19, 1069–1080. [Google Scholar] [CrossRef]
- Fechner, U.; Franke, L.; Renner, S.; Schneider, P.; Schneider, G. Comparison of correlation vector methods for ligand-based similarity searching. J. -Comput.-Aided Mol. Des. 2003, 17, 687–698. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Cao, D.; Liang, Y.; Xu, Q.; Yun, Y.; Li, H. Toward better QSAR/QSPR modeling: Simultaneous outlier detection and variable selection using distribution of model features. J.-Comput.-Aided Mol. Des. 2011, 25, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Insolia, L.; Kenney, A.; Chiaromonte, F.; Felici, G. Simultaneous feature selection and outlier detection with optimality guarantees. Biometrics 2022, 78, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Menjoge, R.S.; Welsch, R.E. A diagnostic method for simultaneous feature selection and outlier identification in linear regression. Comput. Stat. Data Anal. 2010, 54, 3181–3193. [Google Scholar] [CrossRef]
- Kim, S.S.; Park, S.H.; Krzanowski, W.J. Simultaneous variable selection and outlier identification in linear regression using the mean-shift outlier model. J. Appl. Stat. 2008, 35, 283–291. [Google Scholar] [CrossRef]
- Jimenez, F.; Lucena-Sanchez, E.; Sanchez, G.; Sciavicco, G. Multi-Objective Evolutionary Simultaneous Feature Selection and Outlier Detection for Regression. IEEE Access 2021, 9, 135675–135688. [Google Scholar] [CrossRef]
- Park, J.S.; Park, C.G.; Lee, K.E. Simultaneous outlier detection and variable selection via difference-based regression model and stochastic search variable selection. Commun. Stat. Appl. Methods 2019, 26, 149–161. [Google Scholar] [CrossRef]
- Wiegand, P.; Pell, R.; Comas, E. Simultaneous variable selection and outlier detection using a robust genetic algorithm. Chemom. Intell. Lab. Syst. 2009, 98, 108–114. [Google Scholar] [CrossRef]
- Tolvi, J. Genetic algorithms for outlier detection and variable selection in linear regression models. Soft Comput. 2004, 8, 527–533. [Google Scholar] [CrossRef]
- Wen, M.; Deng, B.C.; Cao, D.S.; Yun, Y.H.; Yang, R.H.; Lu, H.M.; Liang, Y.Z. The model adaptive space shrinkage (MASS) approach: A new method for simultaneous variable selection and outlier detection based on model population analysis. Analyst 2016, 141, 5586–5597. [Google Scholar] [CrossRef]
- t-SNE: The Effect of Various Perplexity Values on the Shape. Available online: https://scikit-learn.org/stable/auto_examples/manifold/plot_t_sne_perplexity.html (accessed on 1 June 2023).
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Yu, P.S. A Comprehensive Survey on Graph Neural Networks. IEEE Trans. Neural Netw. Learn. Syst. 2021, 32, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Jegelka, S.; Hu, W.; Leskovec, J. How powerful are graph neural networks? In Proceedings of the 7th International Conference on Learning Representations, ICLR 2019, New Orleans, LA, USA, 6–9 May 2019; pp. 1–17. [Google Scholar]
- Jiang, D.; Wu, Z.; Hsieh, C.Y.; Chen, G.; Liao, B.; Wang, Z.; Shen, C.; Cao, D.; Wu, J.; Hou, T. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminform. 2021, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]























| Configuration | Methods | Thresholds |
|---|---|---|
| 1. Effect of X-outlier elimination | iForest, RF confidence, tSNE2D/kNN | Elimination of 0%, 10%, 30%, 50% of the molecules with the highest anomaly scores |
| 2. Effect of Model-outlier elimination | Standardized residuals | Elimination of the molecules with absolute standardized residuals above 1, 2, 3, 5 and 10 |
| 3. Effect of X-outlier and Model-outlier elimination | Layouts: 1-Simultaneous elimination 2-Model-outlier then X-outlier 3-X-outlier then Model-outlier (RF confidence for X-outlier, Standardized residuals for Model-outlier) | RF confidence: 150 kJ/mol for enthalpy, 50 J/mol/K for entropy. Standardized residuals: 2 kJ/mol or J/mol/K. |
| 4. Effect of y-outlier elimination | IQR | Elimination of the molecules with y-values outside the thresholds of and |
| Property | Outliers | Mol. | Desc. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Elimination | Train | Test | Train | Test | Train | Test | |||
| H | Layout 3, with y-outliers | 1531 | 2506 | 0.998 | 0.995 | 9.51 | 11.88 | 12.72 | 19.41 |
| Layout 3, without y-outliers | 1525 | 2506 | 0.998 | 0.996 | 9.39 | 11.69 | 12.61 | 17.56 | |
| S | Layout 3, with y-outliers | 1514 | 2479 | 0.996 | 0.995 | 7.49 | 8.19 | 9.90 | 11.40 |
| Layout 3, without y-outliers | 1431 | 2479 | 0.991 | 0.988 | 7.56 | 8.28 | 10.00 | 11.49 |
| Configuration | Outlier | Dimensionality | Mol. | Desc. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Elimination | Reduction | Train | Test | Train | Test | Train | Test | |||
| A | No | No | 1785 | 2506 | 0.997 | 0.978 | 16.08 | 26.96 | 30.90 | 71.76 |
| B | Yes | No | 1531 | 2506 | 0.998 | 0.995 | 9.51 | 11.88 | 12.72 | 19.41 |
| C | No | Yes | 1785 | 100 | 0.995 | 0.978 | 15.45 | 24.16 | 36.90 | 70.77 |
| D | Yes | Yes | 1531 | 100 | 0.998 | 0.994 | 9.27 | 11.89 | 14.09 | 21.50 |
| Configuration | Outlier | Dimensionality | Mol. | Desc. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Elimination | Reduction | Train | Test | Train | Test | Train | Test | |||
| A | No | No | 1747 | 2479 | 0.983 | 0.970 | 14.87 | 18.71 | 26.94 | 35.17 |
| B | Yes | No | 1514 | 2479 | 0.996 | 0.995 | 7.49 | 8.19 | 9.90 | 11.40 |
| C | No | Yes | 1747 | 100 | 0.980 | 0.969 | 14.09 | 17.37 | 29.23 | 35.49 |
| D | Yes | Yes | 1514 | 100 | 0.996 | 0.995 | 7.09 | 8.01 | 9.78 | 11.49 |
| Property | Category of Species | Split 1 | Split 2 | Split 3 | Split 4 | Split 5 |
|---|---|---|---|---|---|---|
| H | A | 1 | 0 | 0 | 1 | 2 |
| B | 7 | 8 | 5 | 4 | 4 | |
| C | 18 | 9 | 13 | 12 | 21 | |
| D | 12 | 16 | 11 | 12 | 9 | |
| S | A | 9 | 6 | 5 | 9 | 11 |
| B | 0 | 0 | 0 | 1 | 1 | |
| C | 26 | 23 | 26 | 23 | 33 | |
| D | 25 | 19 | 20 | 20 | 24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinh, C.; Lasala, S.; Herbinet, O.; Meimaroglou, D. On the Development of Descriptor-Based Machine Learning Models for Thermodynamic Properties: Part 2—Applicability Domain and Outliers. Algorithms 2023, 16, 573. https://doi.org/10.3390/a16120573
Trinh C, Lasala S, Herbinet O, Meimaroglou D. On the Development of Descriptor-Based Machine Learning Models for Thermodynamic Properties: Part 2—Applicability Domain and Outliers. Algorithms. 2023; 16(12):573. https://doi.org/10.3390/a16120573
Chicago/Turabian StyleTrinh, Cindy, Silvia Lasala, Olivier Herbinet, and Dimitrios Meimaroglou. 2023. "On the Development of Descriptor-Based Machine Learning Models for Thermodynamic Properties: Part 2—Applicability Domain and Outliers" Algorithms 16, no. 12: 573. https://doi.org/10.3390/a16120573
APA StyleTrinh, C., Lasala, S., Herbinet, O., & Meimaroglou, D. (2023). On the Development of Descriptor-Based Machine Learning Models for Thermodynamic Properties: Part 2—Applicability Domain and Outliers. Algorithms, 16(12), 573. https://doi.org/10.3390/a16120573