
Progressive Multiple Alignment of Graphs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Theory
2.1. Abstract Graph Alignments
2.2. Ambiguous Edges
2.3. Characterization of Pairwise Graph Alignments
2.4. Progressive Alignments
2.5. Labels
2.6. Labels and Scores for Graph Alignments
3. Algorithmic Considerations
3.1. Construction of the Guide Tree Based on Graph Kernels
3.2. Computing Optimal Common Induced Subgraphs
| Algorithm 1: Iterative_Trimming |
 |
| Algorithm 2: VF2_step |
 |
| Algorithm 3: candidate_matches |
Data: Graphs and a matching Result: Set P of match candidates for extending M set of unmatched vertices in set of unmatched vertices in // the anchor is contained in M and would be excluded from the candidates return
P |
3.3. Syntactic and Semantic Compatibility
- (a)
- the two vertex labels and the two vertex labels are compatible and
- (b)
- for all one of the following conditions is satisfied:
- (i)
- and and the two edge labels and are are compatible, or
- (ii)
- and , or
- (iii)
- or .
- (i’)
- If and are connected by directed edges, then iff and the labels of these directed edges, and , are compatible.
- (1)
- If and , or and , then ;
- (2)
- If and , or and , then ;
- (3)
- If and , then .
3.4. Visualization of Alignment Objects
4. Computational Results
4.1. Implementation
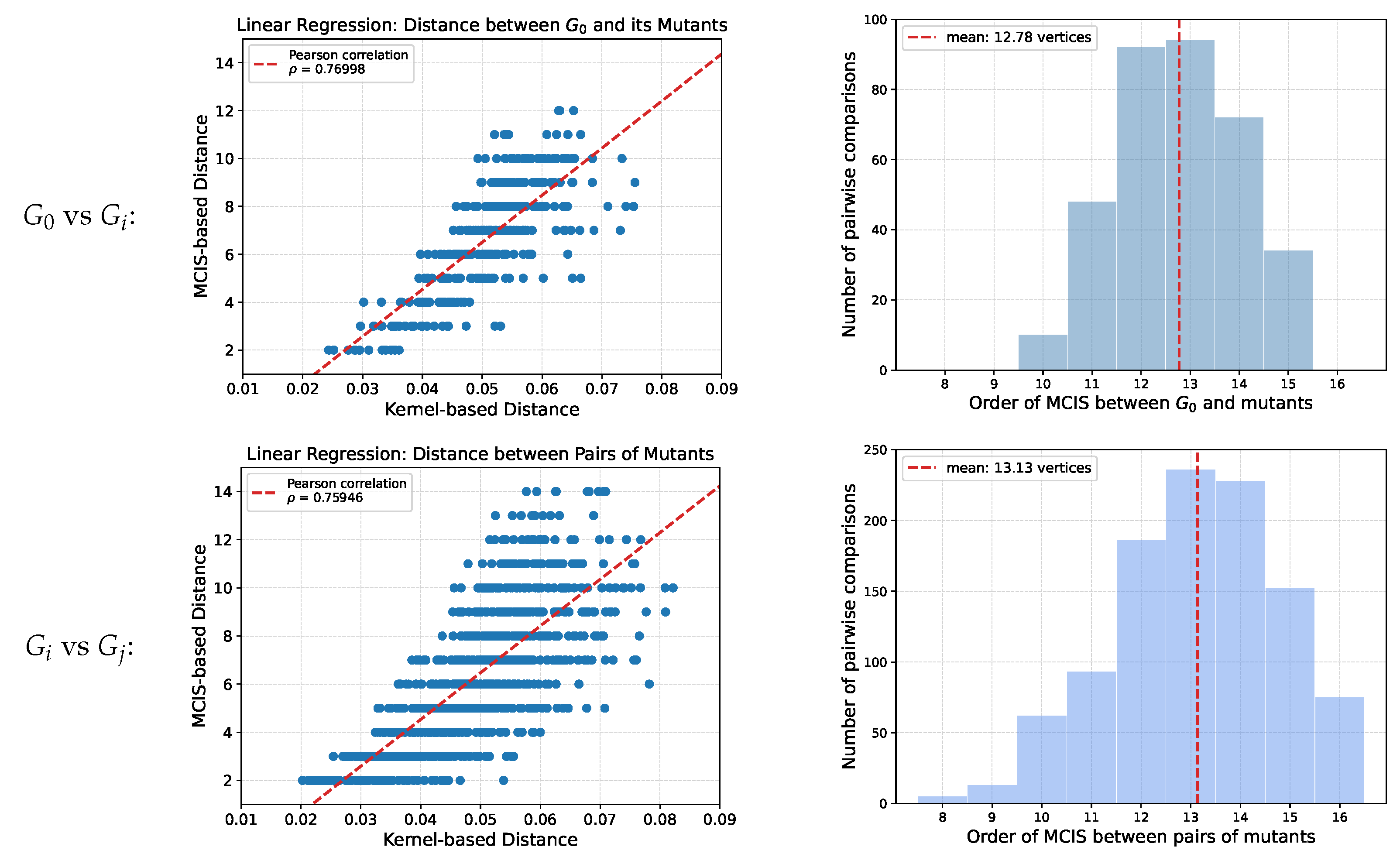
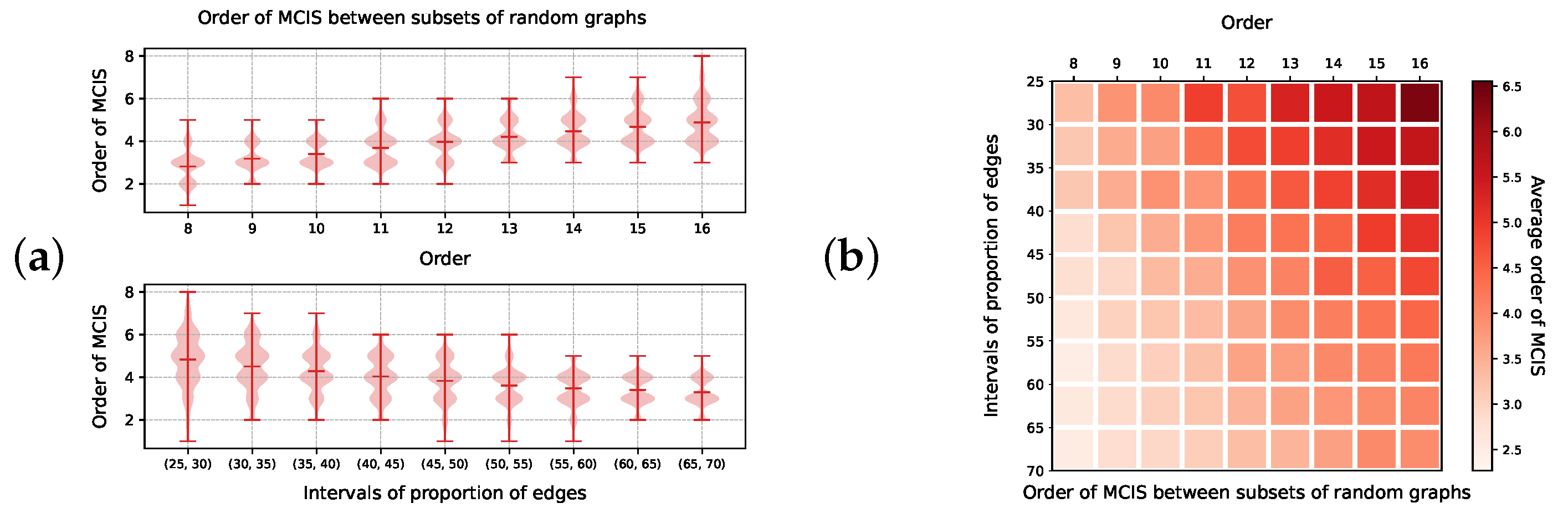
4.2. Generation of Test Sets
4.3. Consensus Graphs
4.4. Ambiguous Edges
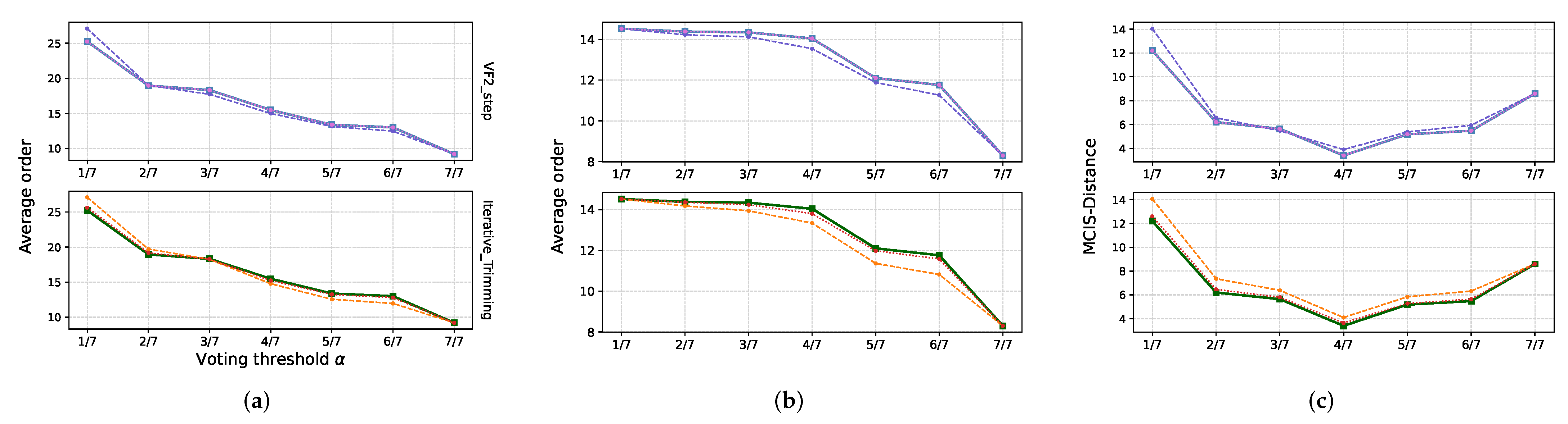
4.5. Running Times
5. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MCIS | maximum common induced subgraph |
| MGA | multiple graph alignment |
Appendix A
Appendix A.1. Additional Information on Graph Similarities

Appendix A.2. Insufficiency of the Standard Version of VF2
| Algorithm A1: candidate_matches_original |
 |

Appendix A.3. Additional Information on Ambiguous Edges

Appendix A.4. Running Times



References
- Rosenberg, M.S. (Ed.) Sequence alignment: Concepts and history. In Sequence Alignment: Methods, Models, Concepts, and Strategies; University of California Press: Oakland, CA, USA, 2009; pp. 1–22. [Google Scholar] [CrossRef]
- Chatzou, M.; Magis, C.; Chang, J.M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple sequence alignment modeling: Methods and applications. Brief. Bioinform. 2015, 17, 1009–1023. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, L.; Zhang, K. Alignment of trees—An alternative to tree edit. Theor. Comput. Sci. 1995, 143, 137–148. [Google Scholar] [CrossRef]
- Höchsmann, M.; Voss, B.; Giegerich, R. Pure multiple RNA secondary structure alignments: A progressive profile approach. Trans. Comput. Biol. Bioinform. 2004, 1, 53–62. [Google Scholar] [CrossRef]
- Berkemer, S.; Höner zu Siederdissen, C.; Stadler, P.F. Compositional properties of alignments. Math. Comput. Sci. 2021, 15, 609–630. [Google Scholar] [CrossRef]
- Berg, J.; Lässig, M. Local graph alignment and motif search in biological networks. Proc. Natl. Acad. Sci. USA 2004, 101, 14689–14694. [Google Scholar] [CrossRef]
- Kuchaiev, O.; Milenković, T.; Memisević, V.; Hayes, W.; Pržulj, N. Topological network alignment uncovers biological function and phylogeny. J. R. Soc. Interface 2010, 7, 1341–1354. [Google Scholar] [CrossRef]
- Mernberger, M.; Klebe, G.; Hüllermeier, E. SEGA: Semiglobal graph alignment for structure-based protein comparison. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Weskamp, N.; Hüllermeier, E.; Kuhn, D.; Klebe, G. Multiple graph alignment for the structural analysis of protein active sites. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xu, J.X.; Berger, B. Global alignment of multiple protein interaction networks with application to functional orthology detection. Proc. Natl. Acad. Sci. USA 2008, 105, 12763–12768. [Google Scholar] [CrossRef]
- Zhang, S.; Tong, H. FINAL: Fast attributed network alignment. In Proceedings of the KDD ’16: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Krishnapuram, B., Shah, M., Smola, A., Aggarwal, C., Shen, D., Eds.; Association for Computing Machinery: New York, NY, USA, 2016; pp. 1345–1354. [Google Scholar] [CrossRef]
- Heimann, M.; Lee, W.; Pan, S.; Chen, K.Y.; Koutra, D. HashAlign: Hash-based alignment of multiple graphs. In Proceedings of the Advances in Knowledge Discovery and Data Mining, PAKDD 2018, Melbourne, VIC, Australia, 3–6 June 2018; Phung, D., Tseng, V., Webb, G., Ho, B., Ganji, M., Rashidi, L., Eds.; Lecture Notes in Computer Science; Springer: Cham, Switzerland, 2018; Volume 10939, pp. 726–739. [Google Scholar] [CrossRef]
- Bayati, M.; Gleich, D.F.; Saberi, A.; Wang, Y. Message-passing algorithms for sparse network alignment. ACM Trans. Knowl. Discov. Data 2013, 7, 3. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, W.; Li, J.; Zhao, K.; Tsung, F.; Li, J. Robust attributed graph alignment via joint structure learning and optimal transport. In Proceedings of the IEEE 39th International Conference on Data Engineering (ICDE), Los Alamitos, CA, USA, 3–7 April 2023; pp. 1638–1651. [Google Scholar] [CrossRef]
- Malmi, E.; Chawla, S.; Gionis, A. Lagrangian relaxations for multiple network alignment. Data Min. Knowl. Discov. 2017, 31, 1331–1358. [Google Scholar] [CrossRef]
- Feng, D.F.; Doolittle, R.F. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol. 1987, 25, 351–360. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, T. On the complexity of multiple sequence alignment. J. Comput. Biol. 1994, 1, 337–348. [Google Scholar] [CrossRef]
- Just, W. Computational complexity of multiple sequence alignment with SP-score. J. Comput. Biol. 2001, 8, 615–623. [Google Scholar] [CrossRef]
- Elias, I. Settling the intractability of multiple alignment. J. Comput. Biol. 2006, 13, 1323–1339. [Google Scholar] [CrossRef]
- Fober, T.; Mernberger, M.; Klebe, G.; Hüllermeier, E. Evolutionary construction of multiple graph alignments for the structural analysis of biomolecules. Bioinformatics 2009, 25, 2110–2117. [Google Scholar] [CrossRef]
- Ngoc, H.T.; Duc, D.D.; Xuan, H.H. A novel ant based algorithm for multiple graph alignment. In Proceedings of the International Conference on Advanced Technologies for Communications (ATC 2014), Hanoi, Vietnam, 15–17 October 2014; Heath, R.W., Quynh, N.X., Lap, L.H., Eds.; IEEE Press: Piscataway, NJ, USA, 2014; pp. 181–186. [Google Scholar] [CrossRef]
- Sokal, R.R.; Michener, C.D. A statistical method for evaluating systematic relationships. Univ. Kansas Sci. Bull. 1958, 38, 1409–1438. [Google Scholar]
- Cordella, L.P.; Foggia, P.; Sansone, C.; Vento, M. A (sub)graph isomorphism algorithm for matching large graphs. IEEE Trans. Pattern Anal. Mach. Intell. 2004, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Alpár Jüttner, A.; Madarasi, P. VF2++—An improved subgraph isomorphism algorithm. Discret. Appl. Math. 2018, 242, 69–81. [Google Scholar] [CrossRef]
- Touzet, H. Comparing similar ordered trees in linear-time. J. Discret. Algorithms 2007, 5, 696–705. [Google Scholar] [CrossRef]
- Stadler, P.F. Alignments of biomolecular contact maps. Interface Focus 2021, 11, 20200066. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, B.; Stoye, J.; Dress, A.W.M. Consistent Equivalence Relations: A Set-Theoretical Framework for Multiple Sequence Alignments; Technical Report; University of Bielefeld, FSPM: Bielefeld, Germany, 1999. [Google Scholar]
- Nelesen, S.; Liu, K.; Zhao, D.; Linder, C.R.; Warnow, T. The effect of the guide tree on multiple sequence alignments and subsequent phylogenetic analyses. In Pacific Sympomsium on Biocomputing PSB’08; Altman, R.B., Dunker, A.K., Hunter, L., Klein, T.E., Eds.; Stanford Univ.: Stanford, CA, USA, 2008; pp. 25–36. [Google Scholar] [CrossRef]
- Zhan, Q.; Ye, Y.; Lam, T.W.; Yiu, S.M.; Wang, Y.; Ting, H.F. Improving multiple sequence alignment by using better guide trees. BMC Bioinform. 2015, 16, S4. [Google Scholar] [CrossRef] [PubMed]
- Bunke, H. On a relation between graph edit distance and maximum common subgraph. Pattern Recognit. Lett. 1997, 18, 689–694. [Google Scholar] [CrossRef]
- Zeng, Z.; Tung, A.K.H.; Wang, J.; Feng, J.; Zhou, L. Comparing stars: On approximating graph edit distance. Proc. VLDB Endow. 2009, 2, 25–36. [Google Scholar] [CrossRef]
- Jia, L.; Gaüzère, B.; Honeine, P. Graph kernels based on linear patterns: Theoretical and experimental comparisons. Expert Syst. Appl. 2022, 189, 116095. [Google Scholar] [CrossRef]
- Schölkopf, B. The kernel trick for distances. In Proceedings of the NIPS’00: Proceedings of the 13th International Conference on Neural Information Processing Systems, Denver, CO, USA, 1 January 2000; Leen, T., Dietterich, T., Tresp, V., Eds.; MIT Press: Cambridge, MA, USA, 2000; pp. 283–289. [Google Scholar]
- Phillips, J.M.; Venkatasubramanian, S. A gentle introduction to the kernel distance. arXiv 2011, arXiv:1103.1625. [Google Scholar] [CrossRef]
- Kriege, N.M.; Johansson, F.D.; Morris, C. A survey on graph kernels. Appl. Netw. Sci. 2020, 5, 6. [Google Scholar] [CrossRef]
- Jia, L.; Gaüzère, B.; Honeine, P. Graphkit-learn: A python library for graph kernels based on linear patterns. Pattern Recognit. Lett. 2021, 143, 113–121. [Google Scholar] [CrossRef]
- Garey, M.R.; Johnson, D.S. Computers and Intractability. A Guide to the Theory of NP Completeness; Freeman: San Francisco, CA, USA, 1979. [Google Scholar]
- Kann, V. On the approximability of the maximum common subgraph problem. In Proceedings of the 9th Annual Symposium on Theoretical Aspects of Computer Science; Cachan, France, 13–15 February 1992; Finkel, A., Jantzen, M., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 1992; Volume 577, pp. 375–388. [Google Scholar] [CrossRef]
- Barrow, H.; Burstall, R. Subgraph isomorphism, matching relational structures and maximal cliques. Inf. Process. Lett. 1976, 4, 83–84. [Google Scholar] [CrossRef]
- McCreesh, C.; Prosser, P.; Trimble, J. A partitioning algorithm for maximum common subgraph problems. In Proceedings of the Twenty-Sixth International Joint Conference on Artificial Intelligence, IJCAI-17, Melbourne, Australia, 19–25 August 2017; Sierra, C., Ed.; AAAI Press: Palo Alto, CA, USA, 2017; pp. 712–719. [Google Scholar] [CrossRef]
- Hoffmann, R.; McCreesh, C.; Reilly, C. Between subgraph isomorphism and maximum common subgraph. In Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017; Markovitch, S., Singh, S., Eds.; AAAI Press: Palo Alto, CA, USA, 2017; Volume 1, pp. 3907–3914. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Li, C.M.; Jiang, H.; He, K. Hybrid learning with new value function for the maximum common induced subgraph problem. In Proceedings of the Thirty-Seventh AAAI Conference on Artificial Intelligence (AAAI-37), Washington, DC, USA, 7–14 February 2023; Williams, B., Chen, Y., Neville, J., Eds.; AAAI Press: Palo Alto, CA, USA, 2023; Volume 4, pp. 4044–4051. [Google Scholar]
- Berezikov, E.; Guryev, V.; Plasterk, R.H.A.; Edwin, C. CONREAL: Conserved regulatory elements anchored alignment algorithm for identification of transcription factor binding sites by phylogenetic footprinting. Genome Res. 2004, 14, 170–178. [Google Scholar] [CrossRef]
- Morgenstern, B.; Prohaska, S.J.; Pohler, D.; Stadler, P.F. Multiple sequence alignment with user-defined anchor points. Algorithms Mol. Biol. 2006, 1, 6. [Google Scholar] [CrossRef]
- Brun, L.; Gaüzère, B.; Fourey, S. Relationships between Graph Edit Distance and Maximal Common Unlabeled Subgraph; Technical Report hal-00714879; HAL: Bangalore, India, 2012. [Google Scholar]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference, Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Vaught, T., Millman, J., Eds.; 2008; pp. 11–15. [Google Scholar]
- González-Laffitte, M.E.; Stadler, P.F. Github Repository of the Progressive Graph Alignment Software ProGrAlign. 2024. Available online: https://github.com/MarcosLaffitte/Progralign (accessed on 23 February 2024).
- Documentation on the Pickle Python Package. Available online: https://docs.python.org/3/library/pickle.html (accessed on 1 March 2024).
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Schneider, T.D. Consensus sequence zen. Appl. Bioinform. 2002, 1, 111–119. [Google Scholar]
- Hagiwara, K.; Edmonson, M.N.; Wheeler, D.A.; Zhang, J. indelPost: Harmonizing ambiguities in simple and complex indel alignments. Bioinformatics 2022, 38, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Giegerich, R. Explaining and controlling ambiguity in dynamic programming. In Proceedings of the Combinatorial Pattern Matching. CPM’00, Montreal, QC, Canada, 21–23 June 2000; Giancarlo, R., Sankoff, D., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2000; Volume 1848. [Google Scholar] [CrossRef]
- Wallace, I.M.; Orla, O.; Higgins, D.G. Evaluation of iterative alignment algorithms for multiple alignment. Bioinformatics 2005, 21, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.H.; Lu, Y.; Wang, Q.W. A polynomial time solvable formulation of multiple sequence alignment. J. Comput. Biol. 2006, 13, 309–319. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González Laffitte, M.E.; Stadler, P.F. Progressive Multiple Alignment of Graphs. Algorithms 2024, 17, 116. https://doi.org/10.3390/a17030116
González Laffitte ME, Stadler PF. Progressive Multiple Alignment of Graphs. Algorithms. 2024; 17(3):116. https://doi.org/10.3390/a17030116
Chicago/Turabian StyleGonzález Laffitte, Marcos E., and Peter F. Stadler. 2024. "Progressive Multiple Alignment of Graphs" Algorithms 17, no. 3: 116. https://doi.org/10.3390/a17030116
APA StyleGonzález Laffitte, M. E., & Stadler, P. F. (2024). Progressive Multiple Alignment of Graphs. Algorithms, 17(3), 116. https://doi.org/10.3390/a17030116