PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales)



Abstract
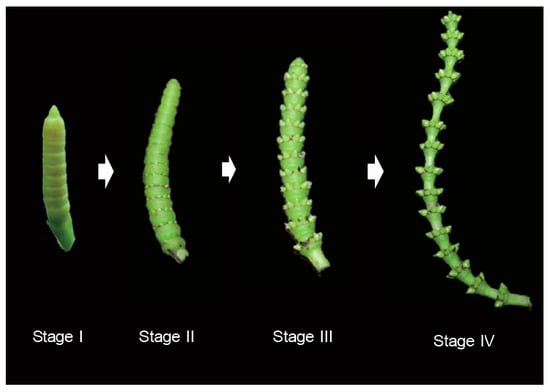
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA Extraction
2.2. cDNA Construction and PacBio SMRT Sequencing
2.3. Illumina Library Construction and Sequencing
2.4. PacBio Long Read Processing and Genome Mapping
2.5. AS and APA Analysis
2.6. Functional Annotation and Classification
2.7. Identification of CDS, Regulatory Proteins and lncRNAs
2.8. Aux/IAA Gene Identification and Sequences Retrieval
2.9. qRT-PCR Validation and Ka/Ks Analysis
2.10. Motif Prediction and Phylogenetic Analysis
3. Results
3.1. Assembly of Full-Length Transcriptome and Genome Mapping
3.2. Functional Annotation and Novel Gene Detection
3.3. AS and APA Analysis
3.4. Identification of CDS, Regulatory Proteins and lncRNAs
3.5. Aux/IAA Gene Identification, qRT-PCR and Adaptive Evolution Analysis
3.6. Motif Prediction and Phylogenetic Analysis
4. Discussion
4.1. Full-Length Transcriptome and Gene Annotation
4.2. AS Analysis
4.3. APA Analysis
4.4. LncRNA Analysis
4.5. Regulator Proteins
4.6. Diversity and Molecular Evolutionary Rates of Aux/IAA Genes
4.7. Phylogenetic Analysis and Structural Detection of Aux/IAA Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Ickert-Bond, S.M.; Renner, S.S. The Gnetales: Recent insights on their morphology, reproductive biology, chromosome numbers, biogeography, and divergence times. J. Syst. Evol. 2015, 54, 1–16. [Google Scholar] [CrossRef]
- Kubitzki, K. General Traits of the Gnetales. In The Families and Genera of Vascular Plants; Kramer, K.U., Green, P.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 378–379. [Google Scholar]
- Endress, P.K. Structure and function of female and bisexual organ complexes in Gnetales. Int. J. Plant Sci. 1996, 157, 113–125. [Google Scholar] [CrossRef]
- Wan, T.; Liu, Z.M.; Li, L.F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.J.; Xin, H.P.; Gong, Y.B.; Liu, Y.; et al. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82–89. [Google Scholar] [CrossRef]
- Bowe, L.M.; Coat, G.; dePamphilis, C.W. Phylogeny of seed plants based on all three genomic compartments: Extant gymnosperms are monophyletic and Gnetales’ closest relatives are conifers. Proc. Natl. Acad. Sci. USA 2000, 97, 4092–4097. [Google Scholar] [CrossRef]
- Donoghue, M.J.; Doyle, J.A. Seed plant phylogeny: Demise of the anthophyte hypothesis? Curr. Biol. 2000, 10, 106–109. [Google Scholar] [CrossRef]
- Doyle, J.A. Seed plant phylogeny and the relationships of Gnetales. Int. J. Plant Sci. 1996, 157, S3–S39. [Google Scholar] [CrossRef]
- Doyle, J.A.; Donoghue, M.J. Seed plant phylogeny and the origin of angiosperms: An experimental cladistic approach. Bot. Rev. 1986, 52, 321–431. [Google Scholar] [CrossRef]
- Kubitzki, K. Gnetaceae. In The Families and Genera of Vascular Plants; Kramer, K.U., Green, P.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 383–386. [Google Scholar]
- Hou, C.; Humphreys, A.M.; Thureborn, O.; Rydin, C. New insights into the evolutionary history of Gnetum (Gnetales). Taxon 2015, 64, 239–253. [Google Scholar] [CrossRef]
- Markgraf, F. Monographie der Gattung Gnetum Ser. 3. Bull. Jard. Bot. Buitenzorg 1930, 10, 407–511. [Google Scholar]
- Hou, C.; Wikström, N.; Strijk, J.; Rydin, C. Resolving phylogenetic relationships and species delimitations in closely related gymnosperms using high-throughput NGS, Sanger sequencing and morphology. Plant Syst. Evol. 2016, 302, 1345–1365. [Google Scholar] [CrossRef]
- Shindo, S.; Ito, M.; Ueda, K.; Kato, M.; Hasebe, M. Characterization of MADS genes in the gymnosperm Gnetum parvifolium and its implication on the evolution of reproductive organs in seed plants. Evol. Dev. 1999, 1, 180–190. [Google Scholar] [CrossRef]
- Winter, K.U.; Becker, A.; Munster, T.; Kim, J.T.; Saedler, H.; Theissen, G. MADS-box genes reveal that gnetophytes are more closely related to conifers than to flowering plants. Proc. Natl. Acad. Sci. USA 1999, 96, 7342–7347. [Google Scholar] [CrossRef]
- Becker, A.; Kaufmann, K.; Freialdenhoven, A.; Vincent, C.; Li, M.A.; Saedler, H.; Theissen, G. A novel MADS-box gene subfamily with a sister-group relationship to class B floral homeotic genes. Mol. Genet. Genomics 2002, 266, 942–950. [Google Scholar]
- Becker, A.; Saedler, H.; Theissen, G. Distinct MADS-box gene expression patterns in the reproductive cones of the gymnosperm Gnetum gnemon. Dev. Genes Evol. 2003, 213, 567–572. [Google Scholar] [CrossRef]
- Becker, A.; Winter, K.U.; Meyer, B.; Saedler, H.; Theissen, G. MADS-box gene diversity in seed plants 300 million years ago. Molec. Biol. Evol. 2000, 17, 1425–1434. [Google Scholar] [CrossRef]
- Lan, Q.; Liu, J.F.; Shi, S.Q.; Deng, N.; Jiang, Z.P.; Chang, E.M. Anatomy, microstructure and endogenous hormone changes in Gnetum parvifolium during anthesis. J. Syst. Evol. 2018, 56, 14–24. [Google Scholar] [CrossRef]
- Woodward, A.W.; Bartel, B. Auxin: Regulation, action, and interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef]
- De Smet, I.; Jürgens, G. Patterning the axis in plants–auxin in control. Curr. Opin. Genet. Dev. 2007, 17, 337–343. [Google Scholar] [CrossRef]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 373–385. [Google Scholar] [CrossRef]
- Guilfoyle, T.J. Aux/IAA proteins and auxin signal transduction. Trends Plant Sci. 1998, 3, 205–207. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.-J.; Zhang, J.-Z. Aux/IAA gene family in plants: Molecular structure, regulation, and function. Int. J. Mol. Sci. 2018, 19, 259. [Google Scholar] [CrossRef]
- Wu, W.T.; Liu, Y.X.; Wang, Y.Q.; Li, H.M.; Liu, J.X.; Tan, J.X.; He, J.D.; Bai, J.W.; Ma, H.L. Evolution analysis of the Aux/IAA gene family in plants shows dual origins and variable nuclear localization signals. Int. J. Mol. Sci. 2017, 18, 2107. [Google Scholar] [CrossRef]
- Abel, S.; Oeller, P.W.; Theologis, A. Early auxin-induced genes encode short-lived nuclear proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 326–330. [Google Scholar] [CrossRef]
- Goldfarb, B.; Lanz-Garcia, C.; Lian, Z.; Whetten, R. Aux/IAA gene family is conserved in the gymnosperm, loblolly pine (Pinus taeda). Tree Physiol. 2003, 23, 1181–1192. [Google Scholar] [CrossRef]
- Jain, M.; Kaur, N.; Garg, R.; Thakur, J.K.; Tyagi, A.K.; Khurana, J.P. Structure and expression analysis of early auxin-responsive Aux/IAA gene family in rice (Oryza sativa). Funct. Integr. Genom. 2006, 6, 47–59. [Google Scholar] [CrossRef]
- Liscum, E.; Reed, J. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 2002, 49, 387–400. [Google Scholar] [CrossRef]
- Wang, Y.J.; Deng, D.X.; Bian, Y.L.; Lv, Y.P.; Xie, Q. Genome-wide analysis of primary auxin-responsive Aux/IAA gene family in maize (Zea mays. L.). Mol. Biol. Rep. 2010, 37, 3991–4001. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715. [Google Scholar] [CrossRef]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Ann. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496. [Google Scholar] [CrossRef]
- Sherstnev, A.; Duc, C.; Cole, C.; Zacharaki, V.; Hornyik, C.; Ozsolak, F.; Milos, P.M.; Barton, G.J.; Simpson, G.G. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat. Struct. Mol. Biol. 2012, 19, 845. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Liu, M.; Downie, B.; Liang, C.; Ji, G.; Li, Q.Q.; Hunt, A.G. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2011, 108, 12533–12538. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Venu, R.; Nobuta, K.; Wu, X.; Notibala, V.; Demirci, C.; Meyers, B.C.; Wang, G.-L.; Ji, G.; Li, Q.Q. Transcriptome dynamics through alternative polyadenylation in developmental and environmental responses in plants revealed by deep sequencing. Genome Res. 2011, 21, 1478–1486. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Chua, N.H. Long noncoding RNA transcriptome of plants. J. Plant Biotechnol. 2015, 13, 319–328. [Google Scholar] [CrossRef]
- Flynn, R.A.; Chang, H.Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Ann. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.-C.; Kean, M.J.; Xu, J.; Chua, N.-H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Boerner, S.; McGinnis, K.M. Computational identification and functional predictions of long noncoding RNA in Zea mays. PLoS ONE 2012, 7, e43047. [Google Scholar] [CrossRef]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.-T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Parker, B.J.; Weiller, G.F. In Silico identification and characterization of mRNA-like noncoding transcripts in Medicago truncatula. Silico Biol. 2007, 7, 485–505. [Google Scholar]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.-Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.X.; Mei, W.B.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT-and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [Green Version]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef] [PubMed]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, 297–299. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, 369–373. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Albert, V.A.; Barbazuk, W.B.; Der, J.P.; Leebens-Mack, J.; Ma, H.; Palmer, J.D.; Rounsley, S.; Sankoff, D.; Schuster, S.C.; Soltis, D.E. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.F.; Banf, M.; Dai, X.B.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Li, J.W.; Ma, W.; Zeng, P.; Wang, J.Y.; Geng, B.; Yang, J.C.; Cui, Q.H. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Froberg, J.E.; Lee, J.T. Long noncoding RNAs: Fresh perspectives into the RNA world. Trends Biochem. Sci. 2014, 39, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Liu, Z.-M.; Liao, Y.-Y.; Yang, M.; Leitch, A.R.; Leitch, I.J.; Van de Peer, Y.; Leebens-Mack, J.H.; Song, C.; Hou, C.; et al. The Welwitschia genome reveals new insights into the early evolutionary history of seed plants. Unpublished.
- Guan, R.; Zhao, Y.P.; Zhang, H.; Fan, G.Y.; Liu, X.; Zhou, W.B.; Shi, C.C.; Wang, J.H.; Liu, W.Q.; Liang, X.M.; et al. Draft genome of the living fossil Ginkgo biloba. Gigascience 2016, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jones, B.; Li, Z.; Frasse, P.; Delalande, C.; Regad, F.; Chaabouni, S.; Latche, A.; Pech, J.-C.; Bouzayen, M. The tomato Aux/IAA transcription factor IAA9 is involved in fruit development and leaf morphogenesis. Plant Cell 2005, 17, 2676–2692. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing Version 3.2.0; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 2 October 2018).
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Hou, C.; Saunders, R.M.; Deng, N.; Wan, T.; Su, Y. Pollination drop proteome and reproductive organ transcriptome comparison in Gnetum reveals entomophilous adaptation. Genes 2019, 10, 800. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Sang, Y.; Liu, X.; Xing, S.; Li, J.; Tang, H.; Sun, L. Transcriptome profile analysis from different sex types of Ginkgo biloba L. Front. Plant. Sci. 2016, 7, 871. [Google Scholar] [CrossRef] [Green Version]
- Pirone-Davies, C.; Prior, N.; von Aderkas, P.; Smith, D.; Hardie, D.; Friedman, W.E.; Mathews, S. Insights from the pollination drop proteome and the ovule transcriptome of Cephalotaxus at the time of pollination drop production. Ann. Bot. 2016, 117, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.-B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [Green Version]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.-K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, G.; Reddy, A. Regulation of alternative splicing of pre-mRNAs by stresses. In Nuclear pre-mRNA Processing in Plants; Reddy, A.S., Golovkin, M., Eds.; Springer: Heidelberg, Germany, 2008; pp. 257–275. [Google Scholar]
- Tai, H.H.; Williams, M.; Iyengar, A.; Yeates, J.; Beardmore, T. Regulation of the β-hydroxyacyl ACP dehydratase gene of Picea mariana by alternative splicing. Plant Cell Rep. 2007, 26, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Cheng, H.; Cai, R.; Li, L.L.; Chang, J.; Zhu, J.; Zhang, F.X.; Chen, L.J.; Wang, Y.; Cheng, S.H. Molecular cloning and function analysis of an anthocyanidin synthase gene from Ginkgo biloba, and its expression in abiotic stress responses. Mol. Cells 2008, 26, 536–547. [Google Scholar] [PubMed]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18. [Google Scholar] [CrossRef]
- Xing, D.; Li, Q.Q. Alternative polyadenylation and gene expression regulation in plants. Wiley Interdiscip. Rev. RNA 2011, 2, 445–458. [Google Scholar] [CrossRef]
- Shen, Y.; Ji, G.; Haas, B.J.; Wu, X.; Zheng, J.; Reese, G.J.; Li, Q.Q. Genome level analysis of rice mRNA 3′-end processing signals and alternative polyadenylation. Nucleic Acids Res. 2008, 36, 3150–3161. [Google Scholar] [CrossRef] [Green Version]
- Chinn, E.; Silverthorne, J.; Hohtola, A. Light-regulated and organ-specific expression of types 1, 2, and 3 light-harvesting complex b mRNAs in Ginkgo biloba. Plant Physiol. 1995, 107, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Simpson, G.G.; Dijkwel, P.P.; Quesada, V.; Henderson, I.; Dean, C. FY is an RNA 3′ end-processing factor that interacts with FCA to control the Arabidopsis floral transition. Cell 2003, 113, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Marquardt, S.; Lister, C.; Swiezewski, S.; Dean, C. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 2010, 327, 94–97. [Google Scholar] [CrossRef]
- Ye, J.; Cheng, S.; Zhou, X.; Chen, Z.; Kim, S.U.; Tan, J.; Zheng, J.; Xu, F.; Zhang, W.; Liao, Y. A global survey of full-length transcriptome of Ginkgo biloba reveals transcript variants involved in flavonoid biosynthesis. Ind. Crops Prod. 2019, 139, 111547. [Google Scholar] [CrossRef]
- Jansson, S.; Gustafsson, P. Type I and type II genes for the chlorophyll a/b-binding protein in the gymnosperm Pinus sylvestris (Scots pine): cDNA cloning and sequence analysis. Plant Mol. Biol. 1990, 14, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.Z.; Shen, G.A.; Liu, C.H.; Liu, X.J.; Tan, F.; Sun, X.F.; Tang, K.X. Molecular cloning and sequence analysis of a novel chalcone synthase cDNA from Ginkgo biloba. DNA Seq. 2004, 15, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Xia, X.; Jiang, H.; Lu, Z.; Cui, J.; Cao, F.; Jin, B. Genome-wide identification and characterization of novel lncRNAs in Ginkgo biloba. Trees 2018, 32, 1429–1442. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Chanderbali, A.S.; Yoo, M.-J.; Zahn, L.M.; Brockington, S.F.; Wall, P.K.; Gitzendanner, M.A.; Albert, V.A.; Leebens-Mack, J.; Altman, N.S.; Ma, H. Conservation and canalization of gene expression during angiosperm diversification accompany the origin and evolution of the flower. Proc. Natl. Acad. Sci. USA 2010, 107, 22570–22575. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Melzer, R.; Theissen, G. Molecular interactions of orthologues of floral homeotic proteins from the gymnosperm Gnetum gnemon provide a clue to the evolutionary origin of ‘floral quartets’. Plant J. 2010, 64, 177–190. [Google Scholar] [CrossRef]
- Hou, C.; Li, L.; Liu, Z.; Su, Y.; Wan, T. Diversity and expression patterns of MADS-box genes in Gnetum luofuense—Implication in functional diversity and evolution. Unpublished.
- Farrona, S.; Hurtado, L.; Bowman, J.L.; Reyes, J.C. The Arabidopsis thaliana SNF2 homolog AtBRM controls shoot development and flowering. Development 2004, 131, 4965–4975. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.X.; Deng, L.L.; Zeng, K.F. Genome-wide in silico identification of membrane-bound transcription factors in plant species. Peerj 2017, 5, e4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.I.; Muthusamy, M.; Nawaz, M.A.; Hong, J.K.; Lim, M.-H.; Kim, J.A.; Jeong, M.-J. Genome-wide analysis of spatiotemporal gene expression patterns during floral organ development in Brassica rapa. Mol. Genet. Genom. 2019, 294, 1403–1420. [Google Scholar] [CrossRef] [PubMed]
- Szecsi, J.; Joly, C.; Bordji, K.; Varaud, E.; Cock, J.M.; Dumas, C.; Bendahmane, M. BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J. 2006, 25, 3912–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heisler, M.G.; Atkinson, A.; Bylstra, Y.H.; Walsh, R.; Smyth, D.R. SPATULA, a gene that controls development of carpel margin tissues in Arabidopsis, encodes a bHLH protein. Development 2001, 128, 1089–1098. [Google Scholar]
- Gish, L.A.; Clark, S.E. The RLK/Pelle family of kinases. Plant J. 2011, 66, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.E.; Running, M.P.; Meyerowitz, E.M. CLAVATA1, a regulator of meristem and flower development in Arabidopsis. Development 1993, 119, 397–418. [Google Scholar]
- Clark, S.E.; Williams, R.W.; Meyerowitz, E.M. The CLAVATA1 gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis. Cell 1997, 89, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Avila, C.; Pérez-Rodríguez, J.; Canovas, F. Molecular characterization of a receptor-like protein kinase gene from pine (Pinus sylvestris L.). Planta 2006, 224, 12–19. [Google Scholar] [CrossRef]
- Ljung, K. Auxin metabolism and homeostasis during plant development. Development 2013, 140, 943–950. [Google Scholar] [CrossRef] [Green Version]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, C.; Deng, N.; Su, Y. PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales). Forests 2019, 10, 1043. https://doi.org/10.3390/f10111043
Hou C, Deng N, Su Y. PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales). Forests. 2019; 10(11):1043. https://doi.org/10.3390/f10111043
Chicago/Turabian StyleHou, Chen, Nan Deng, and Yingjuan Su. 2019. "PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales)" Forests 10, no. 11: 1043. https://doi.org/10.3390/f10111043
APA StyleHou, C., Deng, N., & Su, Y. (2019). PacBio Long-Read Sequencing Reveals the Transcriptomic Complexity and Aux/IAA Gene Evolution in Gnetum (Gnetales). Forests, 10(11), 1043. https://doi.org/10.3390/f10111043