High Genetic Diversity and Low Differentiation in Michelia shiluensis, an Endangered Magnolia Species in South China

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. Primers and Fragment Amplification
2.3. Data Analysis
2.3.1. nSSR Analysis
2.3.2. cpDNA Analysis
3. Results
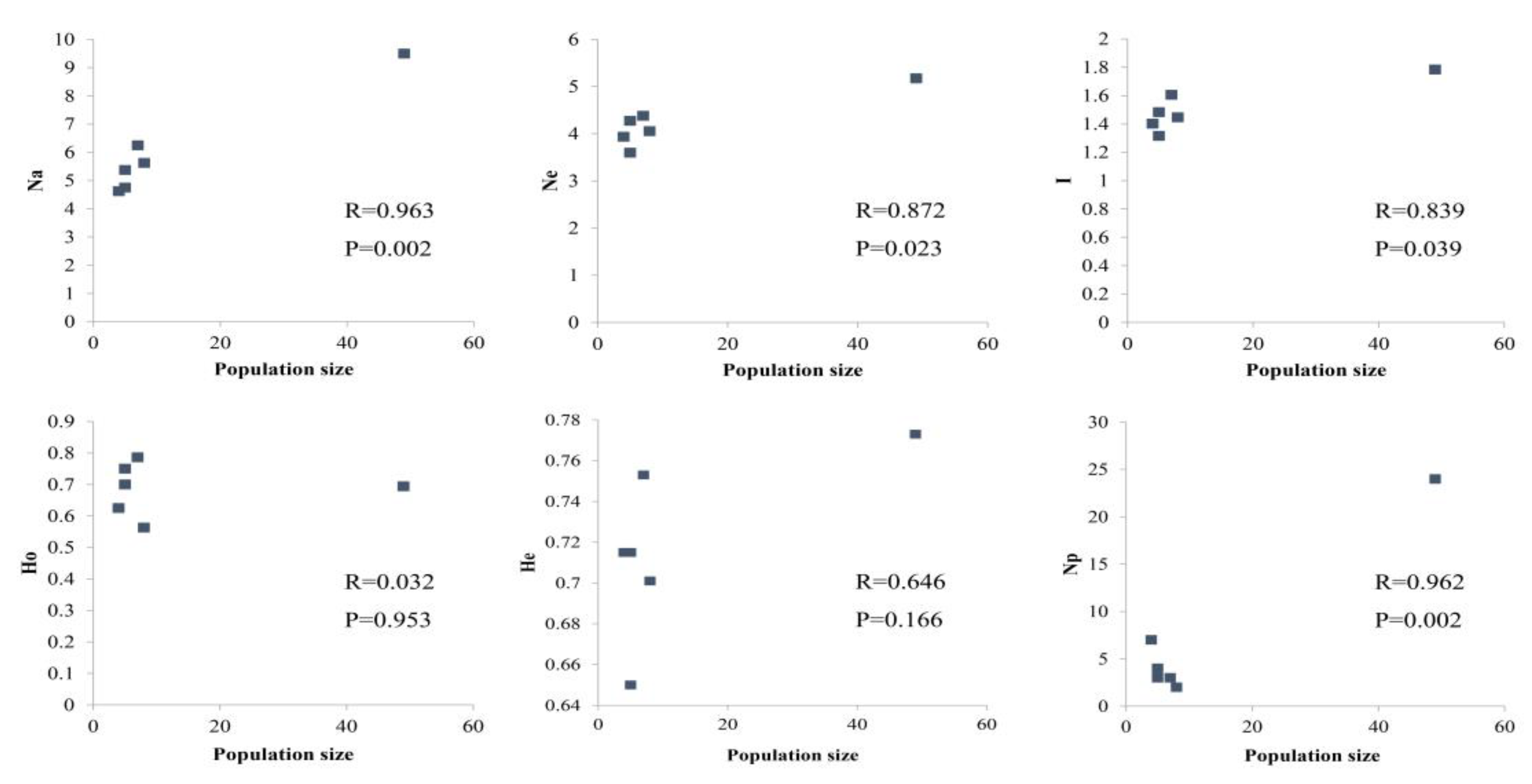
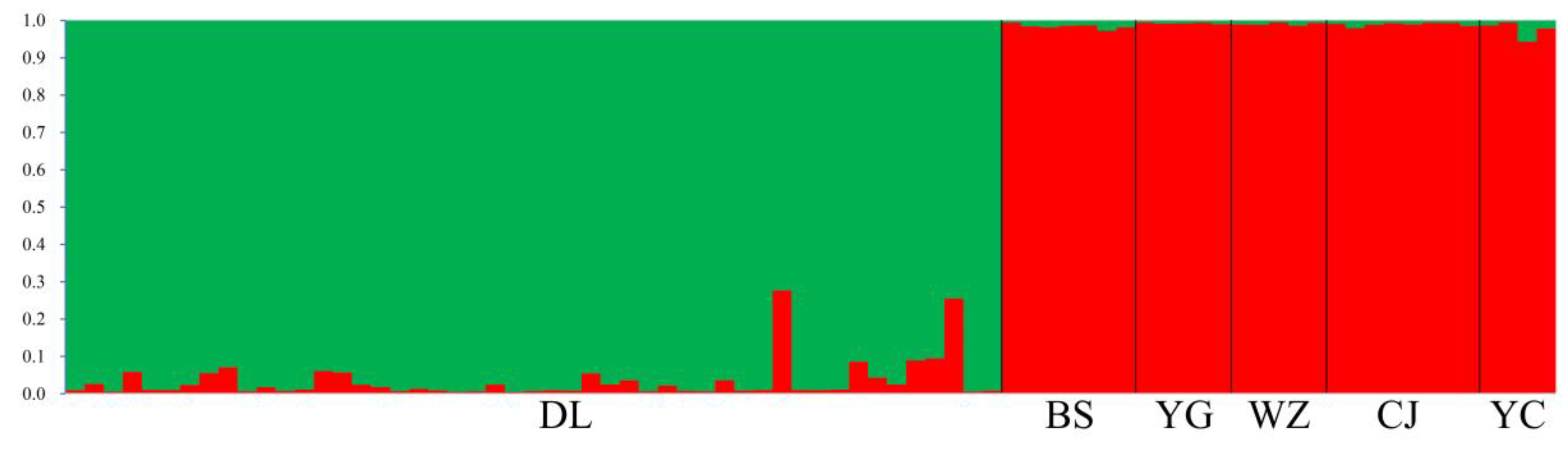
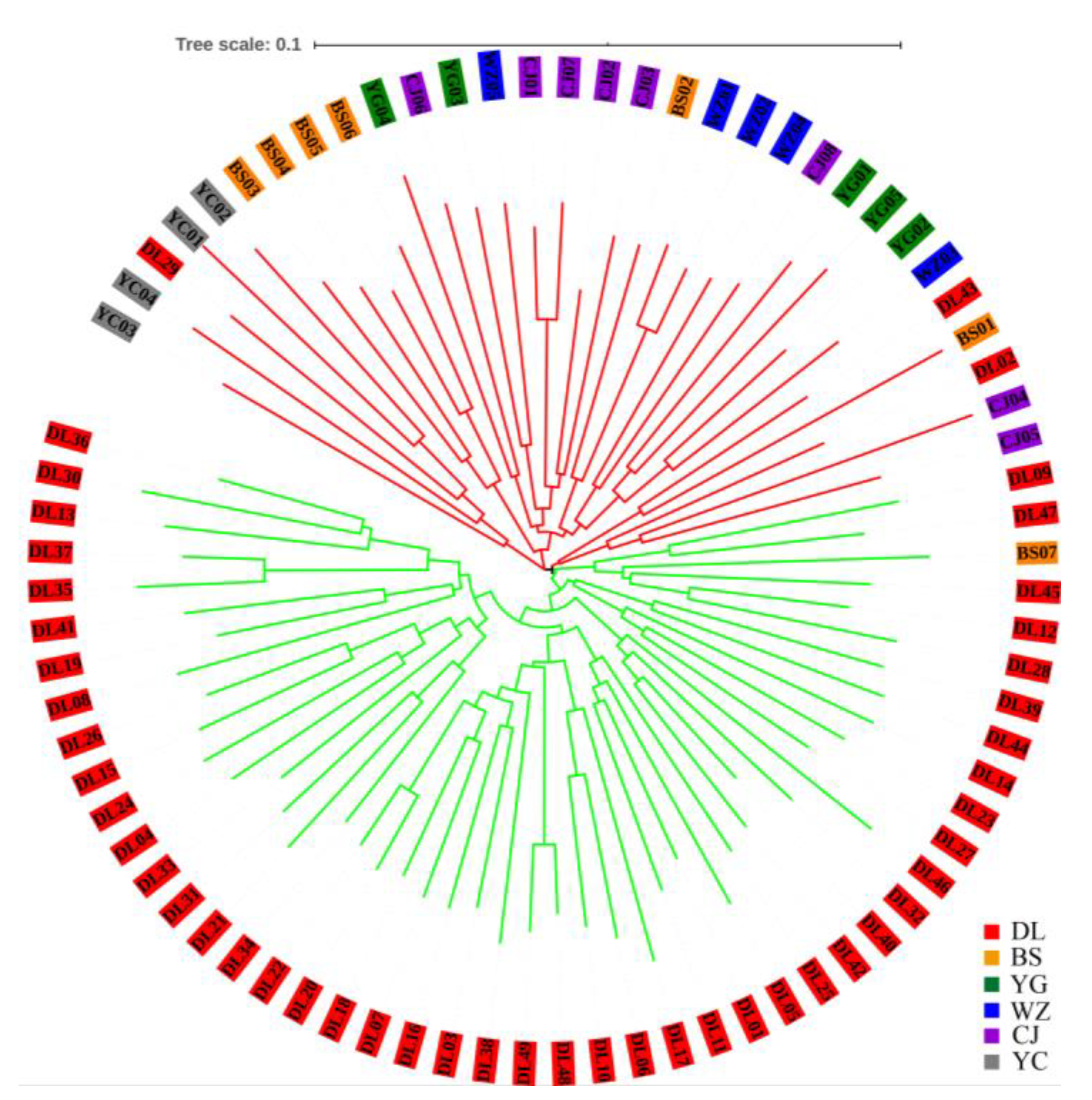
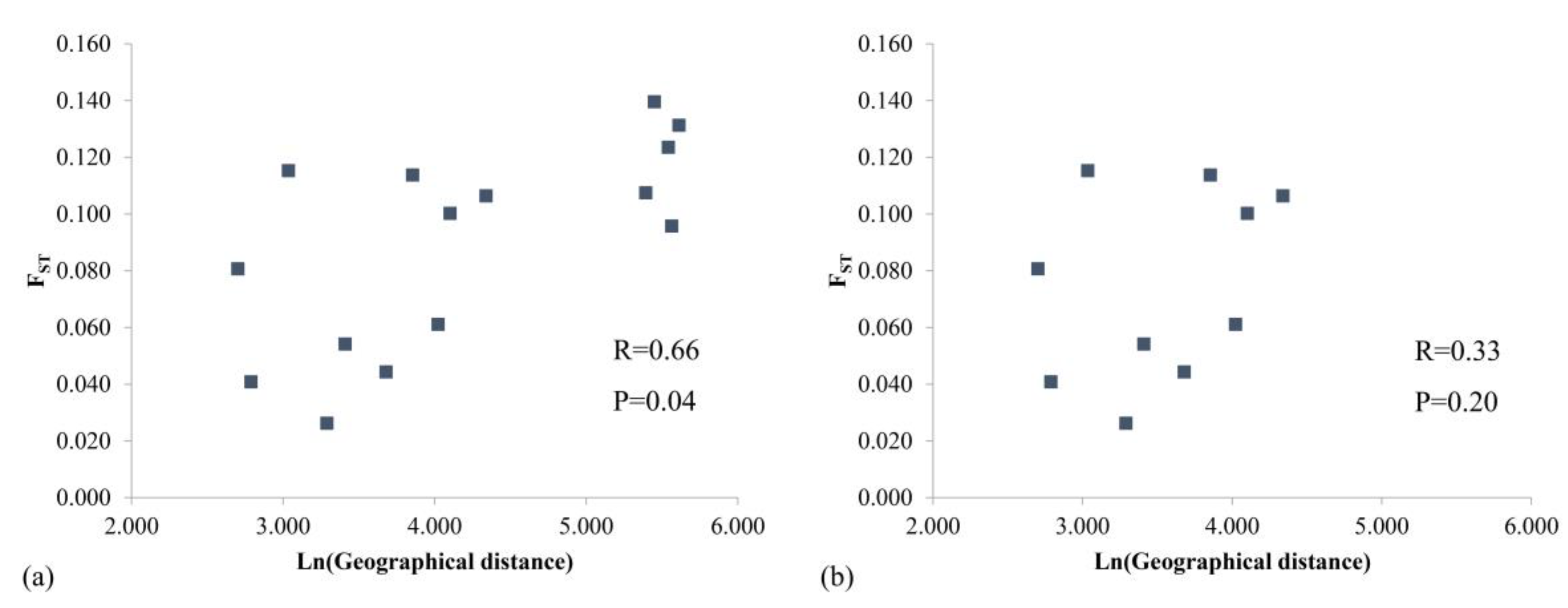
3.1. Genetic Diversity and Population Structure from nSSR Markers
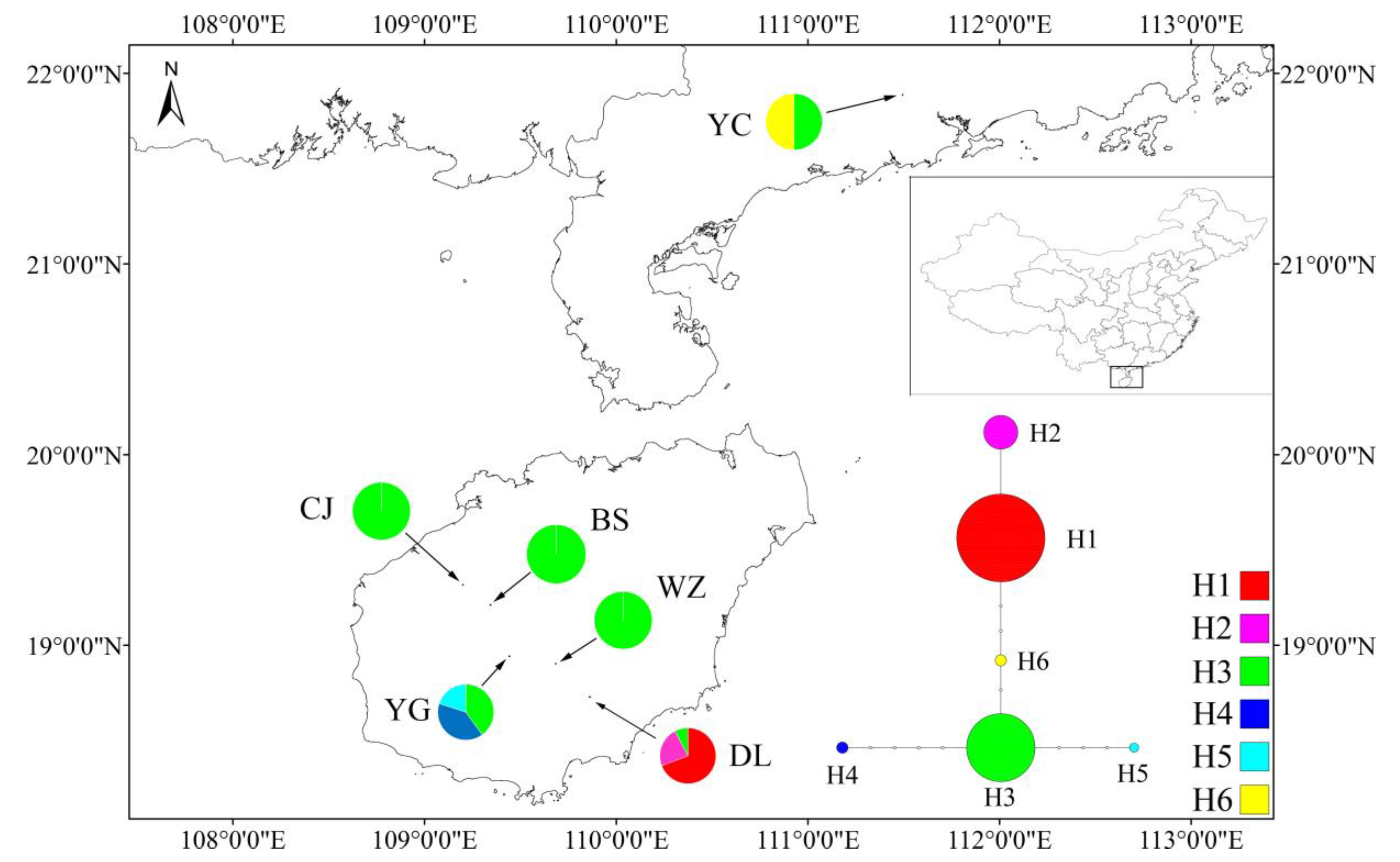
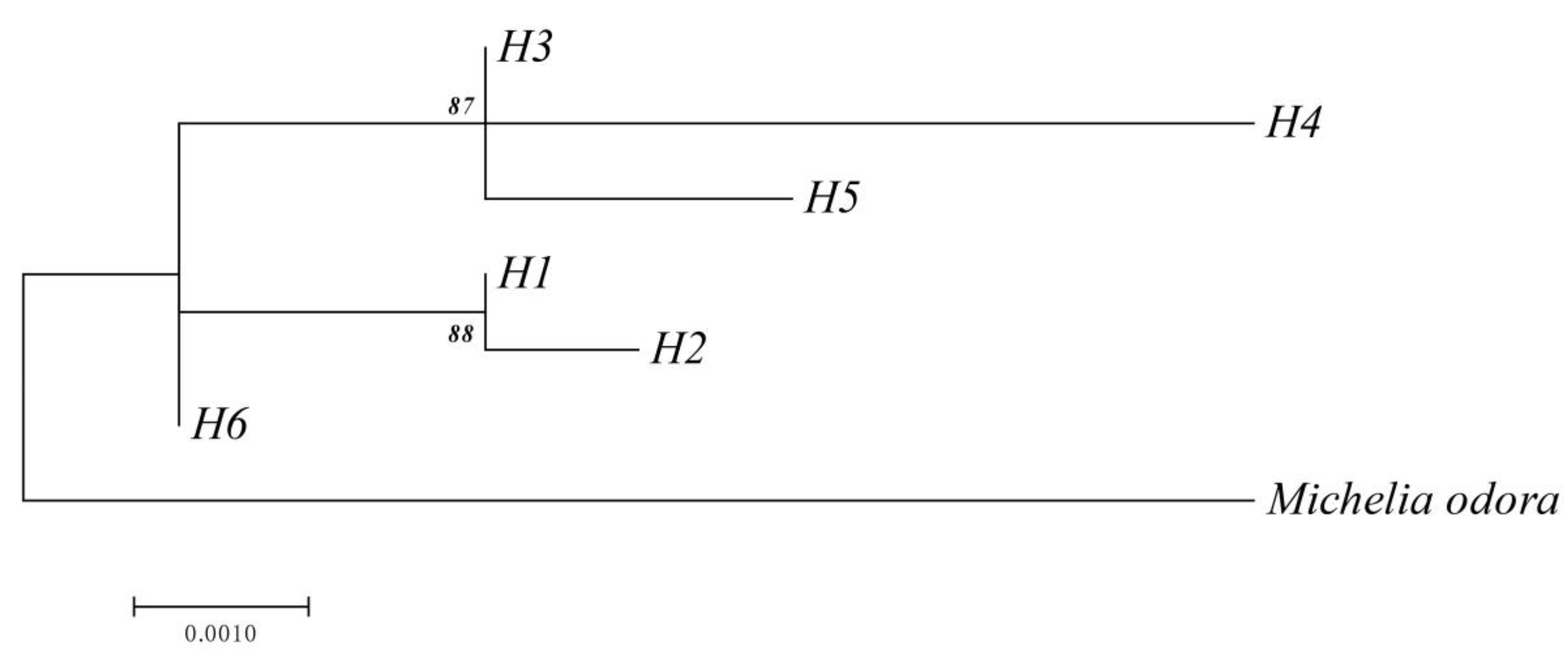
3.2. Genetic Diversity and Population Structure from cpDNA Marker
4. Discussion
4.1. High Level of Genetic Diversity
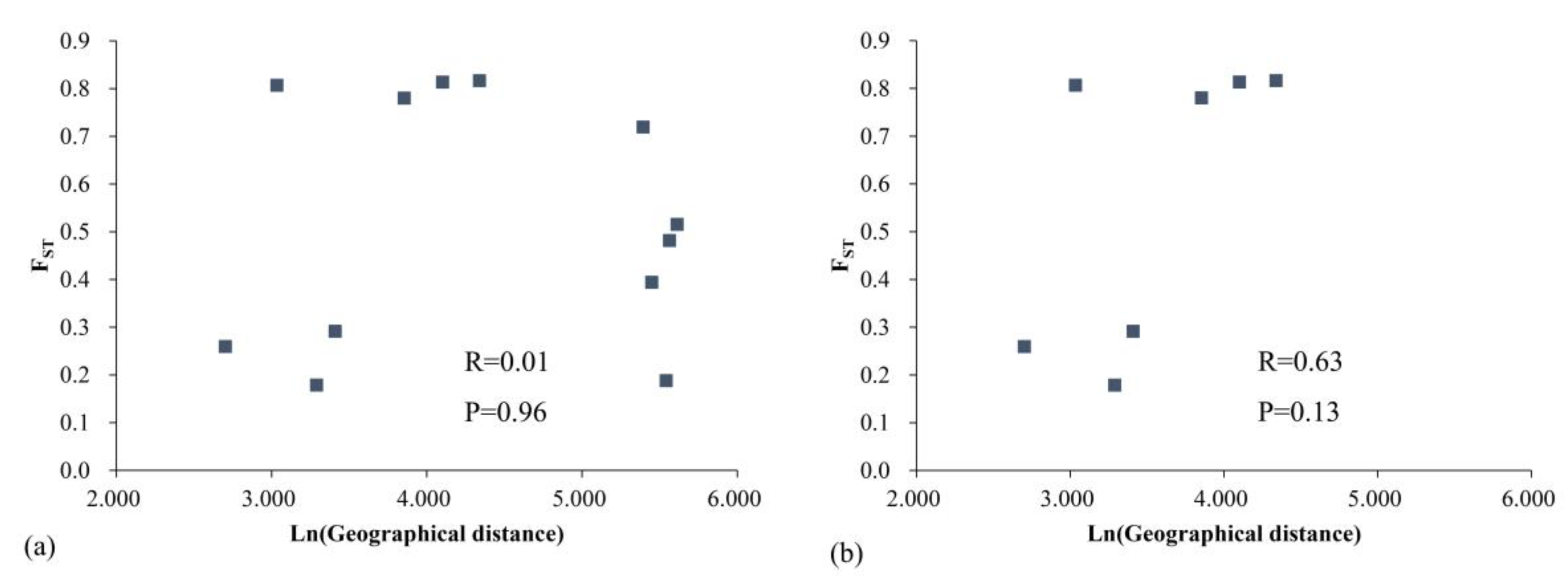
4.2. Lack of Genetic Differentiation between Populations
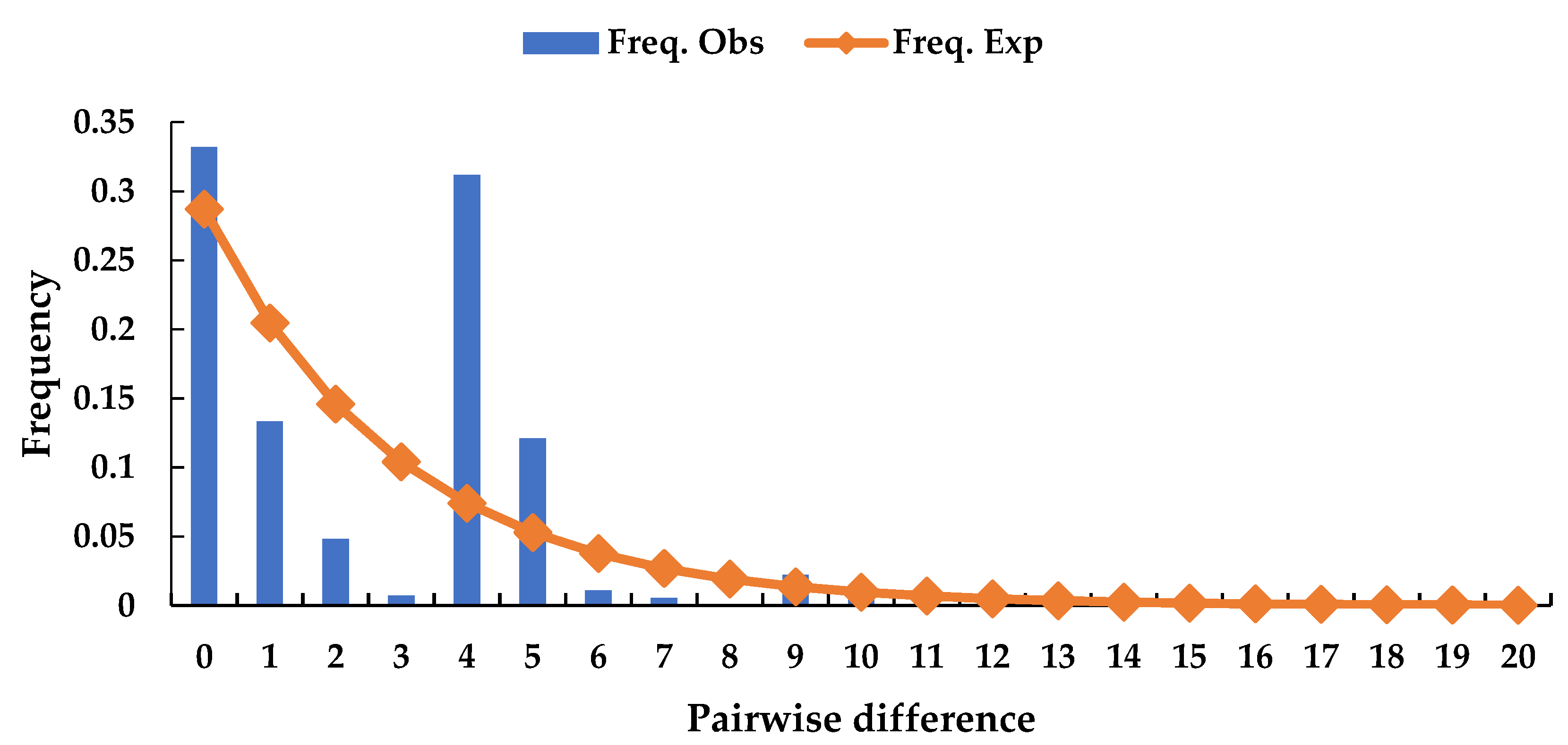
4.3. Demographic History and Population Structure
4.4. Implication for Conservation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tamaki, I.; Setsuko, S.; Tomaru, N. Genetic variation and differentiation in populations of a threatened tree, Magnolia stellata: Factors influencing the level of within-population genetic variation. Heredity 2008, 100, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.B.; Ma, K.P. Geographic distribution patterns and status assessment of threatened plants in China. Biodivers. Conserv. 2008, 17, 1783–1798. [Google Scholar] [CrossRef]
- Caballero, A.; Bravo, I.; Wang, J. Inbreeding load and purging: Implications for the short-term survival and the conservation management of small populations. Heredity 2017, 118, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lande, R. Anthropogenic, ecological and genetic factors in extinction and conservation. Popul. Ecol. 1998, 40, 259–269. [Google Scholar] [CrossRef]
- Tang, C.Q.; Yang, Y.; Ohsawa, M.; Momohara, A.; Hara, M.; Cheng, S.; Fan, S. Population structure of relict Metasequoia glyptostroboides and its habitat fragmentation and degradation in south-central China. Biol. Conserv. 2011, 144, 279–289. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1996, 351, 1291–1298. [Google Scholar]
- Loveless, M.D.; Hamrick, J.L. Ecological determinants of genetic structure in plant populations. Annu. Rev. Ecol. Syst. 1984, 15, 65–95. [Google Scholar] [CrossRef]
- Schoen, D.J.; Morgan, M.T.; Bataillon, T. How does self-pollination evolve? Inferences from floral ecology and molecular genetic variation. Philos. Trans. R. Soc. Lond. B 1996, 351, 1281–1290. [Google Scholar]
- Setsuko, S.; Ishida, K.; Ueno, S.; Tsumura, Y.; Tomaru, N. Population differentiation and gene flow within a metapopulation of a threatened tree, Magnolia stellata (Magnoliaceae). Am. J. Bot. 2007, 94, 128–136. [Google Scholar] [CrossRef]
- Hanski, I.; Simberloff, D. The metapopulation approach, its history, conceptual domain, and application to conservation. In Metapopulation Biology; Elsevier: Amsterdam, The Netherlands, 1997; pp. 5–26. [Google Scholar]
- Ellstrand, N.C.; Elam, D.R. Population genetic consequences of small population size: Implications for plant conservation. Annu. Rev. Ecol. Syst. 1993, 24, 217–242. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Bao, Z.X.; Qu, Y.; Li, W.; Li, Z.Z. Genetic diversity and population structure of the medicinal orchid Gastrodia elata revealed by microsatellite analysis. Biochem. Syst. Ecol. 2014, 54, 182–189. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and conservation biology. C. R. Biol. 2003, 326, 22–29. [Google Scholar] [CrossRef]
- Kingston, N.; Waldren, S.; Smyth, N. Conservation genetics and ecology of Angiopteris chauliodonta Copel. (Marattiaceae), a critically endangered fern from Pitcairn Island, South Central Pacific Ocean. Biol. Conserv. 2004, 117, 309–319. [Google Scholar] [CrossRef]
- Lande, R.; Barrowdough, G. Effective population size, genetic variation, and their use in population management. In Viable Populations for Conservation; Soule, M.E., Ed.; Cambridge University Press: Cambridge, UK, 1987; p. 87. [Google Scholar]
- Godt, M.J.W.; Caplow, F.; Hamrick, J. Allozyme diversity in the federally threatened golden paintbrush, Castilleja levisecta (Scrophulariaceae). Conserv. Genet. 2005, 6, 87–99. [Google Scholar] [CrossRef]
- Luan, S.; Chiang, T.Y.; Gong, X. High genetic diversity vs. low genetic differentiation in Nouelia insignis (Asteraceae), a narrowly distributed and endemic species in China, revealed by ISSR fingerprinting. Ann. Bot. 2006, 98, 583–589. [Google Scholar] [CrossRef]
- Cicuzza, D.; Newton, A.; Oldfield, S. The Red List of Magnoliaceae; Botanic Gardens Conservation International: London, UK, 2007. [Google Scholar]
- Francisco-Ortega, J.; Wang, F.G.; Wang, Z.S.; Xing, F.W.; Liu, H.; Xu, H.; Xu, W.X.; Luo, Y.B.; Song, X.Q.; Gale, S.; et al. Endemic seed plant species from Hainan Island: A checklist. Bot. Rev. 2010, 76, 295–345. [Google Scholar] [CrossRef]
- Wang, F.; Ye, H.; Zhao, N. Studies on the spermatophytic flora of E’ huangzhang Nature Reserve in Yangchun of Guangdong Province. Guangxi Zhiwu 2003, 23, 495–504. [Google Scholar]
- Deng, Y.; Luo, Y.; He, Y.; Qin, X.; Li, C.; Deng, X. Complete chloroplast genome of Michelia shiluensis and a comparative analysis with four Magnoliaceae species. Forests 2020, 11, 267. [Google Scholar] [CrossRef] [Green Version]
- Yi, R.; Chen, Y.; Han, J.; Hu, Q.; Li, H.; Wu, H. Identification and biological characteristics of Diaporthe ueckerae causing dieback disease on Michelia shiluensis. Sci. Silvae Sin. 2018, 54, 80–88. [Google Scholar]
- Wei, Y.; Hong, F.; Yuan, L.; Kong, Y.; Shi, Y. Population distribution and age structure characteristics of Michelia shiluensis, an endangered and endemic species in Hainan Island. Chin. J. Trop. Crops 2017, 38, 2280–2284. [Google Scholar]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Sahu, J.; Sen, P.; Choudhury, M.D.; Barooah, M.; Modi, M.K.; Talukdar, A.D. Towards an efficient computational mining approach to identify EST-SSR markers. Bioinformation 2012, 8, 201–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavov, G.T.; Howe, G.T.; Gyaourova, A.V.; Birkes, D.S.; Adams, W.T. Estimating pollen flow using SSR markers and paternity exclusion: Accounting for mistyping. Mol. Ecol. 2005, 14, 3109–3121. [Google Scholar] [CrossRef]
- Ueno, S.; Setsuko, S.; Kawahara, T.; Yoshimaru, H. Genetic diversity and differentiation of the endangered Japanese endemic tree Magnolia stellata using nuclear and chloroplast microsatellite markers. Conserv. Genet. 2005, 6, 563–574. [Google Scholar] [CrossRef]
- Birky, C.W., Jr.; Fuerst, P.; Maruyama, T. Organelle gene diversity under migration, mutation, and drift: Equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to nuclear genes. Genetics 1989, 121, 613–627. [Google Scholar]
- Birky, C.W., Jr. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef] [Green Version]
- Petit, R.J.; Pineau, E.; Demesure, B.; Bacilieri, R.; Ducousso, A.; Kremer, A. Chloroplast DNA footprints of postglacial recolonization by oaks. Proc. Natl. Acad. Sci. USA 1997, 94, 9996–10001. [Google Scholar] [CrossRef] [Green Version]
- Forcioli, D.; Saumitou-Laprade, P.; Valero, M.; Vernet, P.; Cuguen, J. Distribution of chloroplast DNA diversity within and among populations in gynodioecious Beta vulgaris ssp. maritima (Chenopodiaceae). Mol. Ecol. 1998, 7, 1193–1204. [Google Scholar] [CrossRef]
- Caron, H.; Dumas, S.; Marque, G.; Messier, C.; Bandou, E.; Petit, R.; Kremer, A. Spatial and temporal distribution of chloroplast DNA polymorphism in a tropical tree species. Mol. Ecol. 2000, 9, 1089–1098. [Google Scholar] [CrossRef]
- Petit, R.J.; El Mousadik, A.; Pons, O. Identifying populations for conservation on the basis of genetic markers. Conserv. Biol. 1998, 12, 844–855. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Holland, M.M.; Parson, W. GeneMarker® HID: A reliable software tool for the analysis of forensic STR data. J. Forensic Sci. 2011, 56, 29–35. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Wen, W. Documentation for STRUCTURE Software: Version 2; University of Chicago: Chicago, IL, USA, 2003. [Google Scholar]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.; Yang, R.C.; Boyle, T.B.J.; Ye, Z.; Xiyan, J.M.; Yang, R.; Boyle, T.J. PopGene32, Microsoft Windows-Based Freeware for Population Genetic Analysis. Version 1.32; University of Alberta: Edmonton, AB, Canada, 2000. [Google Scholar]
- Schneider, S.D.; Roessli, D.; Excoffier, L. Arlequin: A software for population genetics data analysis. User Man. Ver 2000, 2, 2496–2497. [Google Scholar]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.C.K.A.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar]
- Pons, O.; Petit, R. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar]
- Verma, S.; Rana, T.S.; Ranade, S.A. Genetic variation and clustering in Murraya paniculata complex as revealed by single primer amplification reaction methods. Curr. Sci. India 2009, 96, 1210–1216. [Google Scholar]
- George, S.; Sharma, J.; Yadon, V.L. Genetic diversity of the endangered and narrow endemic Piperia yadonii (Orchidaceae) assessed with ISSR polymorphisms. Am. J. Bot. 2009, 96, 2022–2030. [Google Scholar] [CrossRef]
- Qiao, Q.; Zhang, C.Q.; Milne, R.I. Population genetics and breeding system of Tupistra pingbianensis (Liliaceae), a naturally rare plant endemic to SW China. J. Syst. Evol. 2010, 48, 47–57. [Google Scholar] [CrossRef]
- Setsuko, S.; Ishida, K.; Tomaru, N. Size distribution and genetic structure in relation to clonal growth within a population of Magnolia tomentosa Thunb. (Magnoliaceae). Mol. Ecol. 2004, 13, 2645–2653. [Google Scholar] [CrossRef]
- Zhao, X.; Ma, Y.; Sun, W.; Wen, X.; Milne, R. High genetic diversity and low differentiation of Michelia coriacea (Magnoliaceae), a critically endangered endemic in southeast Yunnan, China. Int. J. Mol. Sci. 2012, 13, 4396–4411. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xiao, A.H.; Ma, L.Y.; Chen, F.J.; Sang, Z.Y.; Duan, J. Identification of Magnolia wufengensis (Magnoliaceae) cultivars using phenotypic traits, SSR and SRAP markers: Insights into breeding and conservation. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef]
- Lázaro-Nogal, A.; Matesanz, S.; García-Fernández, A.; Traveset, A.; Valladares, F. Population size, center–periphery, and seed dispersers’ effects on the genetic diversity and population structure of the Mediterranean relict shrub Cneorum tricoccon. Ecol. Evol. 2017, 7, 7231–7242. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, A.; Rymer, P.; Holford, P.; Morris, E.C.; Renshaw, A. Evidence for clonality, breeding system, genetic diversity and genetic structure in large and small populations of Melaleuca deanei (Myrtaceae). Aust. J. Bot. 2019, 67, 36–45. [Google Scholar] [CrossRef]
- Tamaki, I.; Setsuko, S.; Tomaru, N. Genetic diversity and structure of remnant Magnolia stellata populations affected by anthropogenic pressures and a conservation strategy for maintaining their current genetic diversity. Conserv. Genet. 2016, 17, 715–725. [Google Scholar] [CrossRef]
- Bezemer, N.; Krauss, S.L.; Roberts, D.G.; Hopper, S.D. Conservation of old individual trees and small populations is integral to maintain species’ genetic diversity of a historically fragmented woody perennial. Mol. Ecol. 2019, 28, 3339–3357. [Google Scholar] [CrossRef]
- Leimu, R.; Mutikainen, P.; Koricheva, J.; Fischer, M. How general are positive relationships between plant population size, fitness and genetic variation? J. Ecol. 2006, 94, 942–952. [Google Scholar] [CrossRef]
- Lu, S.Y.; Hong, K.H.; Liu, S.L.; Cheng, Y.P.; Wu, W.L.; Chiang, T.Y. Genetic variation and population differentiation of Michelia formosana (Magnoliaceae) based on cpDNA variation and RAPD fingerprints: Relevance to post-Pleistocene recolonization. J. Plant Res. 2002, 115, 203–216. [Google Scholar] [CrossRef]
- Azuma, H.; Thien, L.B.; Kawano, S. Molecular phylogeny of Magnolia (Magnoliaceae) inferred from cpDNA sequences and evolutionary divergence of the floral scents. J. Plant Res. 1999, 112, 291–306. [Google Scholar] [CrossRef]
- Frankham, R.; Briscoe, D.A.; Ballou, J.D. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Yu, H.H.; Yang, Z.L.; Sun, B.; Liu, R.N. Genetic diversity and relationship of endangered plant Magnolia officinalis (Magnoliaceae) assessed with ISSR polymorphisms. Biochem. Syst. Ecol. 2011, 39, 71–78. [Google Scholar] [CrossRef]
- Young, A.G.; Merriam, H.G.; Warwick, S.I. The effects of forest fragmentation on genetic variation in Acer saccharum Marsh. (sugar maple) populations. Heredity 1993, 71, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.; Jin, X.; Cai, M. Pollination biology of Michelia crassipes YW Law. In Proceedings of the II International Symposium on Germplasm of Ornamentals, Atlanta, GA, USA, 8–12 August 2016; International Society for Horticultural Science: Leuven, Belgium, 2017; pp. 297–304. [Google Scholar]
- Zhao, X.; Sun, W. Abnormalities in sexual development and pollinator limitation in Michelia coriacea (Magnoliaceae), a critically endangered endemic to Southeast Yunnan, China. Flora 2009, 204, 463–470. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, S.; Wu, F.; Wang, Y. Inferring genetic variation and demographic history of Michelia yunnanensis Franch. (Magnoliaceae) from chloroplast DNA sequences and microsatellite markers. Front. Plant Sci. 2017, 8, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Higashi, H.; Mitsui, Y.; Setoguchi, H. Distinct phylogeographic structures of wild radish (Raphanus sativus L. var. raphanistroides Makino) in Japan. PLoS ONE 2015, 10, e0135132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. Evolution in Mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar]
- Tian, K.; Zhang, G.; Cheng, X.; He, S.; Yang, Y.; Yang, Y. The habitat fragility of Manglietiastrum sinicum. Acta Bot. Yunnanica 2003, 25, 551–556. [Google Scholar]
- Allendorf, F.W. Isolation, gene flow and genetic differentiation among populations. In Genetics and Conservation: A Reference for Managing Wild Animal and Plant Populations; Schoenwald-Cox, C.M., Chambers, S.M., Thomas, W.L., Eds.; Menlo Park/Benjamin Cummings: San Francisco, CA, USA, 1983; pp. 51–65. [Google Scholar]
- Moyle, L.C. Correlates of genetic differentiation and isolation by distance in 17 congeneric Silene species. Mol. Ecol. 2006, 15, 1067–1081. [Google Scholar] [CrossRef]
- Holsinger, K.E.; Gottlieb, L.; Falk, D.A. Conservation of rare and endangered plants: Principles and prospects. In Genetics and Conservation of Rare Plants; Falk, D.A., Holsinger, K.E., Eds.; Oxford University Press: Oxford, UK, 1991; pp. 195–208. [Google Scholar]
- Kawecki, T.J.; Ebert, D. Conceptual issues in local adaptation. Ecol. Lett. 2004, 7, 1225–1241. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Kohn, B.; Spencer, S.; Guo, X.; Li, Y.; Yang, X.; Shi, H.; Gleadow, A. Cenozoic denudation history of southern Hainan Island, South China Sea: Constraints from low temperature thermochronology. Tectonophysics 2011, 504, 100–115. [Google Scholar] [CrossRef]
- Liang, G. A study of the genesis of Hainan Island. Geol. China 2018, 45, 693–705. [Google Scholar]
- Zhou, T.H.; Dong, S.S.; Li, S.; Zhao, G.F. Genetic structure within and among populations of Saruma henryi, an endangered plant endemic to China. Biochem. Genet. 2012, 50, 146–158. [Google Scholar] [CrossRef]
- Ge, X.J.; Zhou, X.L.; Li, Z.C.; Hsu, T.W.; Schaal, B.A.; Chiang, T.Y. Low genetic diversity and significant population structuring in the relict Amentotaxus argotaenia complex (Taxaceae) based on ISSR fingerprinting. J. Plant Res. 2005, 118, 415–422. [Google Scholar] [CrossRef]
- Li, J.M.; Jin, Z.X. Genetic structure of endangered Emmenopterys henryi Oliv. based on ISSR polymorphism and implications for its conservation. Genetica 2008, 133, 227–234. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop | N | Na ± SE | Ne ± SE | I ± SE | Ho ± SE | He ± SE | Np | FIS ± SE | PPL (%) |
|---|---|---|---|---|---|---|---|---|---|
| DL | 49 | 9.500 ± 1.195 | 5.174 ± 0.824 | 1.784 ± 0.137 | 0.694 ± 0.033 | 0.773 ± 0.035 | 24 | 0.093 ± 0.050 | 100.00 |
| BS | 7 | 6.250 ± 0.526 | 4.380 ± 0.470 | 1.606 ± 0.095 | 0.786 ± 0.071 | 0.753 ± 0.026 | 3 | -0.044 ± 0.092 | 100.00 |
| YG | 5 | 5.375 ± 0.596 | 4.275 ± 0.619 | 1.483 ± 0.151 | 0.750 ± 0.098 | 0.715 ± 0.058 | 4 | -0.054 ± 0.105 | 100.00 |
| WZ | 5 | 4.750 ± 0.648 | 3.598 ± 0.416 | 1.316 ± 0.197 | 0.700 ± 0.120 | 0.650 ± 0.094 | 3 | −0.080 ± 0.096 | 87.50 |
| CJ | 8 | 5.625 ± 0.885 | 4.053 ± 0.614 | 1.447 ± 0.172 | 0.563 ± 0.085 | 0.701 ± 0.054 | 2 | 0.207 ± 0.099 | 100.00 |
| YC | 4 | 4.625 ± 0.324 | 3.936 ± 0.399 | 1.402 ± 0.109 | 0.625 ± 0.082 | 0.715 ± 0.046 | 7 | 0.095 ± 0.127 | 100.00 |
| Mean | 13 | 6.021 ± 0.376 | 4.236 ± 0.233 | 1.506 ± 0.061 | 0.686 ± 0.035 | 0.718 ± 0.023 | 7.17 | 0.039 ± 0.040 | 97.92 |
| Overall | 78 | 14.375 ± 1.647 | 7.019 ± 0.971 | 2.120 ± 0.153 | 0.689 ± 0.025 | 0.825 ± 0.039 | - | 0.156 ± 0.033 | 100.00 |
| Markers | Source of Variation | Degree of Freedom | Sum of Squares | Variance Component | Percentage of Variation | p Value |
|---|---|---|---|---|---|---|
| nSSR | Among populations | 5 | 47.296 | 0.353 | 10.18 | <0.01 |
| Within populations | 150 | 467.370 | 3.116 | 89.92 | <0.01 | |
| Total | 155 | 514.667 | 3.469 | 100 | ||
| cpDNA | Among populations | 5 | 66.612 | 1.437 | 77.56 | <0.01 |
| Within populations | 72 | 29.927 | 0.416 | 22.44 | <0.01 | |
| Total | 77 | 96.539 | 1.853 | 100 |
| Haplotype | H1 | H2 | H3 | H4 | H5 | H6 | Sum | Hd | Pi | |
|---|---|---|---|---|---|---|---|---|---|---|
| Pops | DL | 34 | 12 | 3 | 0 | 0 | 0 | 49 | 0.464 | 0.00074 |
| BS | 0 | 0 | 7 | 0 | 0 | 0 | 7 | 0 | 0 | |
| YG | 0 | 0 | 2 | 2 | 1 | 0 | 5 | 0.800 | 0.00333 | |
| WZ | 0 | 0 | 5 | 0 | 0 | 0 | 5 | 0 | 0 | |
| CJ | 0 | 0 | 8 | 0 | 0 | 0 | 8 | 0 | 0 | |
| YC | 0 | 0 | 2 | 0 | 0 | 2 | 4 | 0.667 | 0.00117 | |
| Overall | 34 | 12 | 27 | 2 | 1 | 2 | 78 | 0.674 | 0.00220 | |
| Hap. pops | Neutrality test | Mismatch distribution | ||
|---|---|---|---|---|
| Tajima’s D(P) | Fu’s FS(P) | SSD (PSSD) | HRag (PHRag) | |
| All pops | 0.075(0.525) | 2.786(0.904) | 0.059(0.25) | 0.190(0.41) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Liu, T.; Xie, Y.; Wei, Y.; Xie, Z.; Shi, Y.; Deng, X. High Genetic Diversity and Low Differentiation in Michelia shiluensis, an Endangered Magnolia Species in South China. Forests 2020, 11, 469. https://doi.org/10.3390/f11040469
Deng Y, Liu T, Xie Y, Wei Y, Xie Z, Shi Y, Deng X. High Genetic Diversity and Low Differentiation in Michelia shiluensis, an Endangered Magnolia Species in South China. Forests. 2020; 11(4):469. https://doi.org/10.3390/f11040469
Chicago/Turabian StyleDeng, Yanwen, Tingting Liu, Yuqing Xie, Yaqing Wei, Zicai Xie, Youhai Shi, and Xiaomei Deng. 2020. "High Genetic Diversity and Low Differentiation in Michelia shiluensis, an Endangered Magnolia Species in South China" Forests 11, no. 4: 469. https://doi.org/10.3390/f11040469
APA StyleDeng, Y., Liu, T., Xie, Y., Wei, Y., Xie, Z., Shi, Y., & Deng, X. (2020). High Genetic Diversity and Low Differentiation in Michelia shiluensis, an Endangered Magnolia Species in South China. Forests, 11(4), 469. https://doi.org/10.3390/f11040469