
Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, RNA Extraction, and Library Construction
2.2. Sequencing, Assembly, and Annotation
2.3. SNP Calling and Differential Analysis
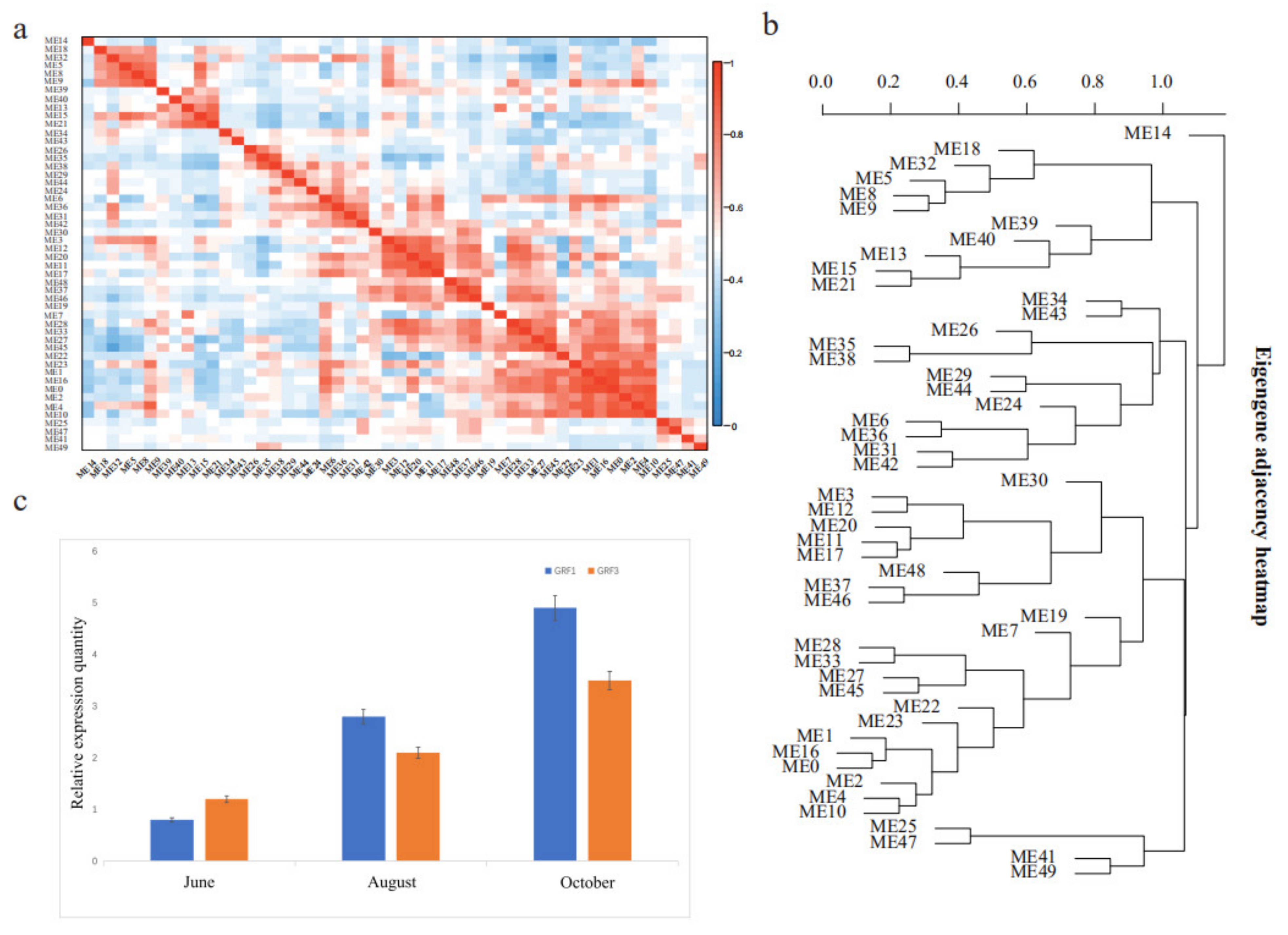
2.4. WGCNA Analysis and Quantitative Real-Time PCR
3. Results and Discussion
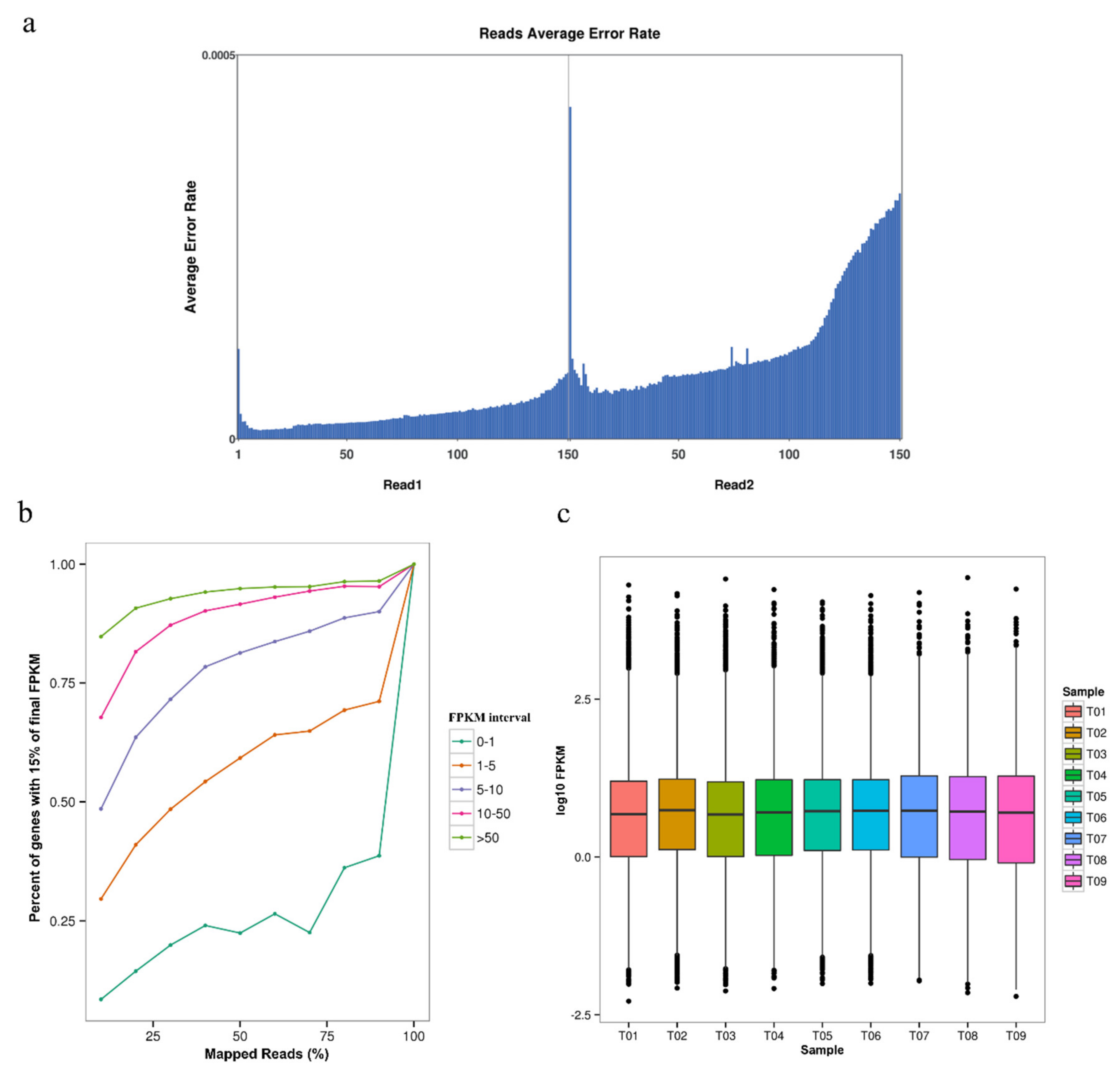
3.1. Overall Characteristics and Quality Evaluation in G. biloba Transcriptome
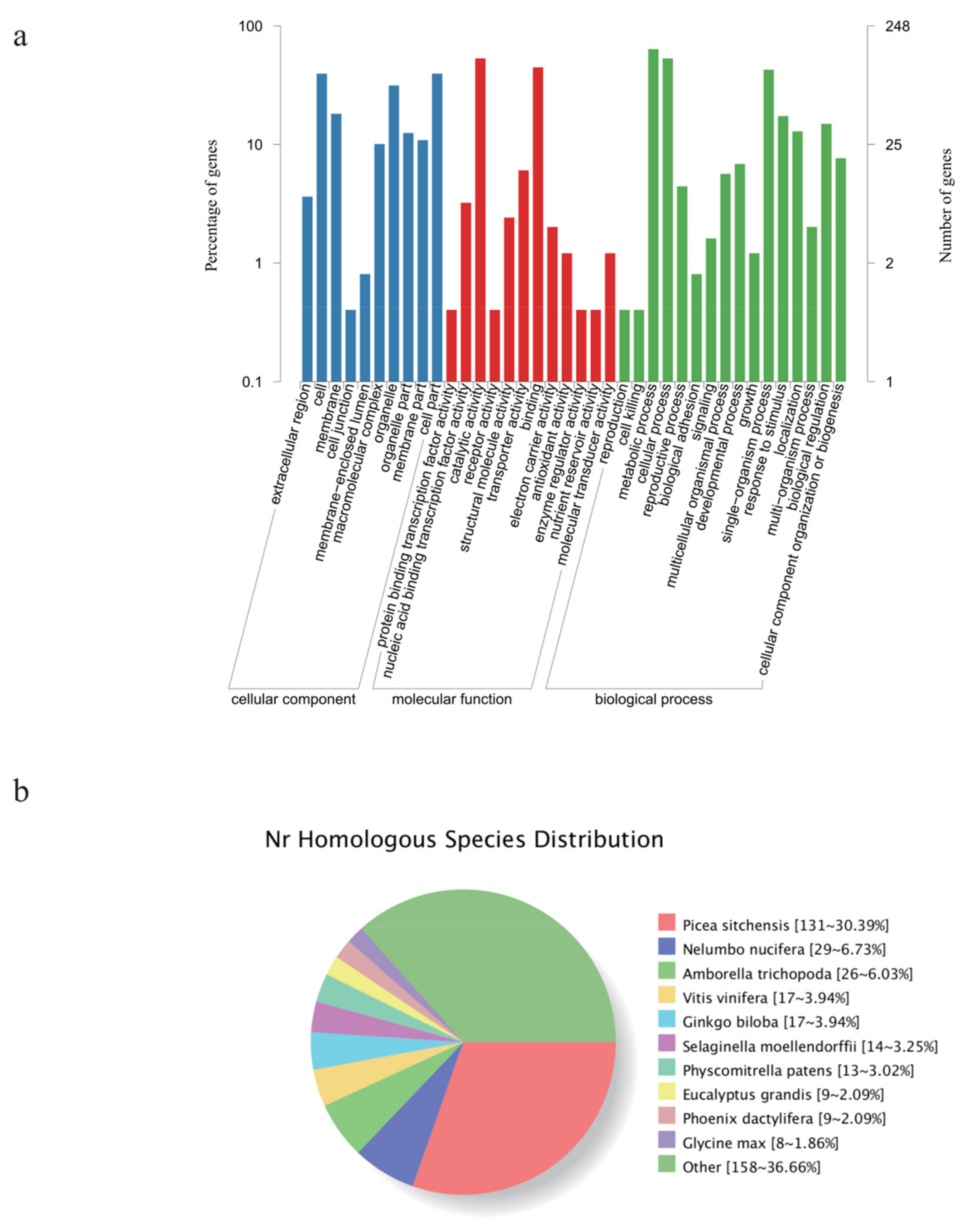
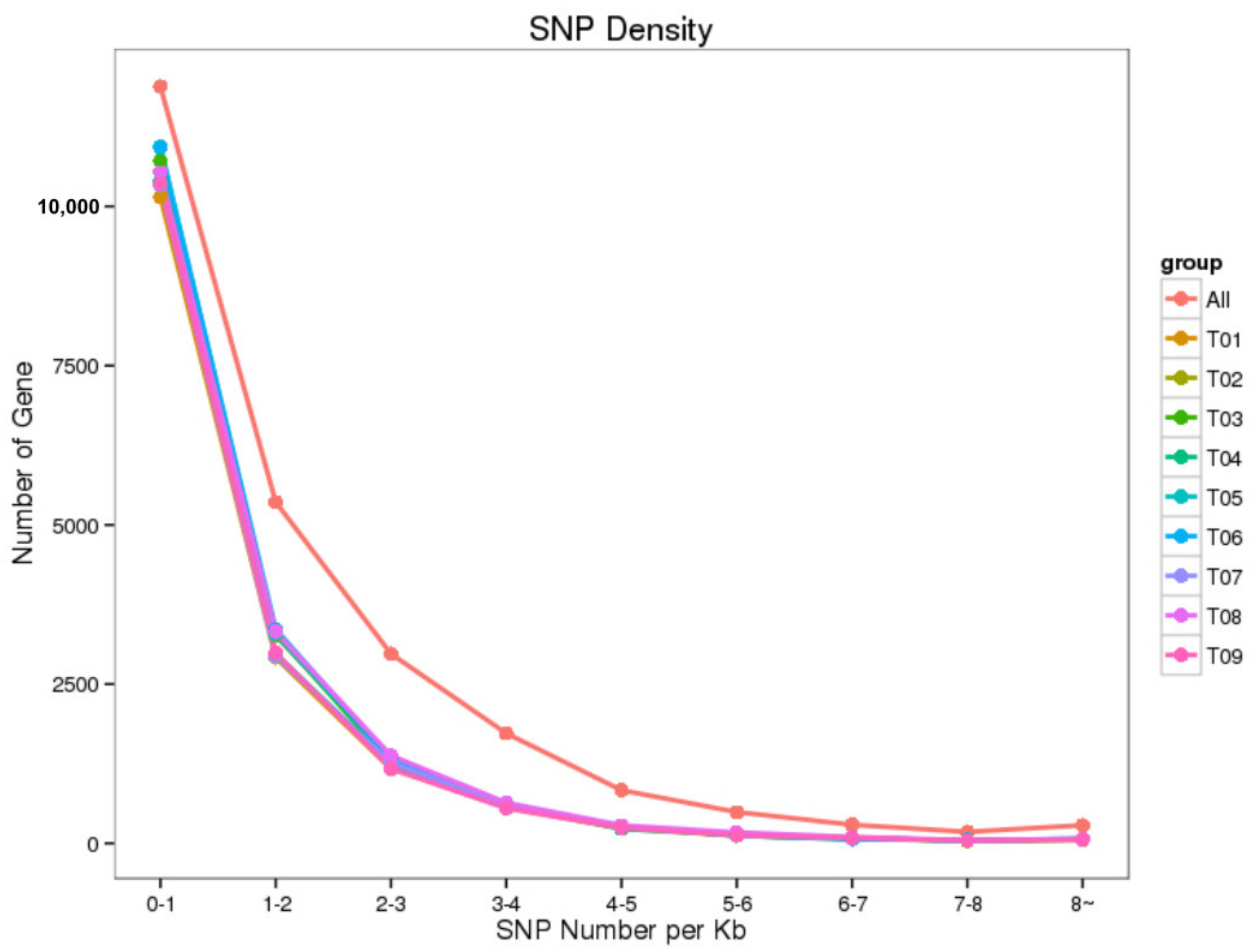
3.2. Identification of New Genes and Potential SNPs
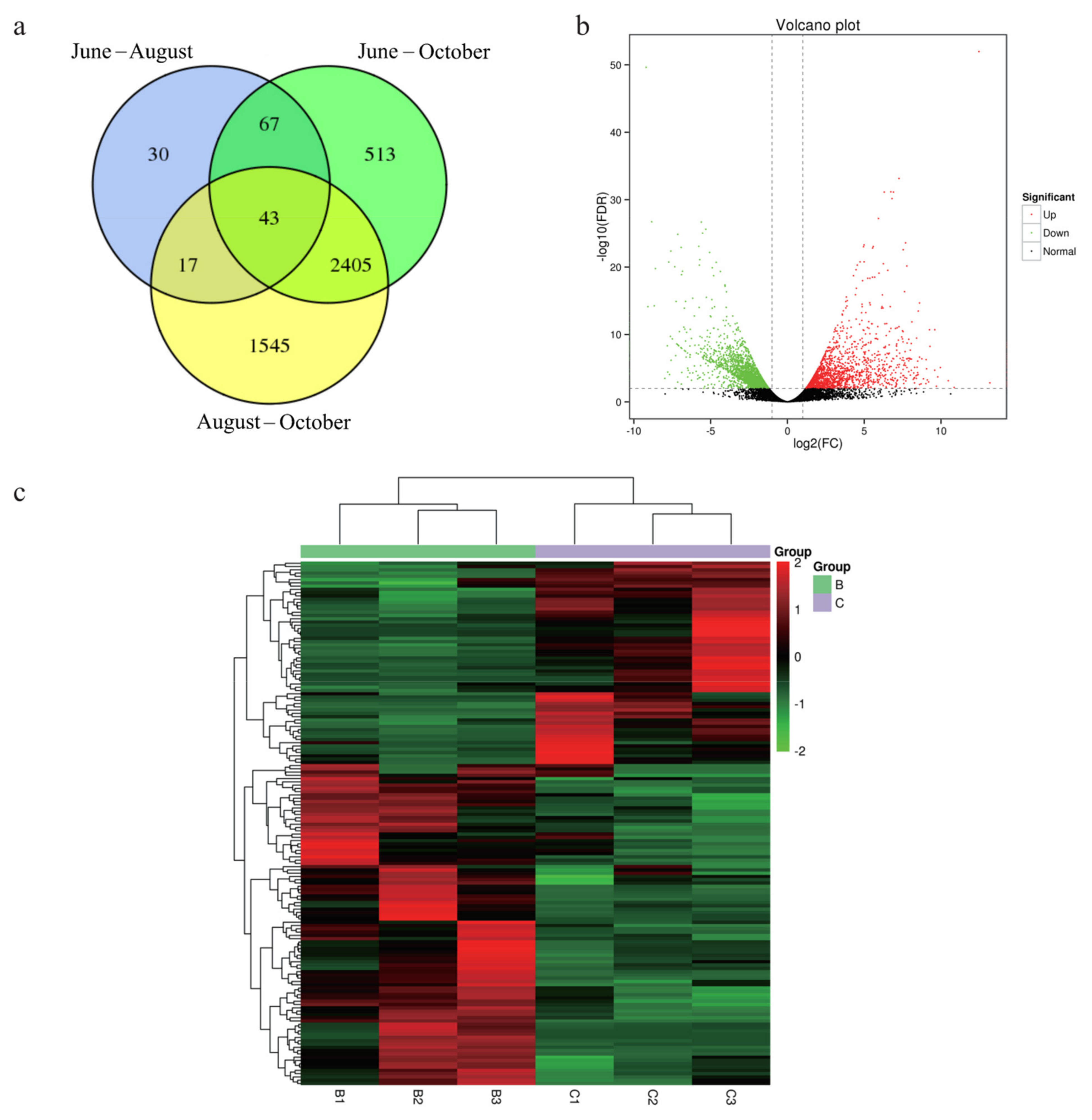
3.3. Differential Expression Analysis
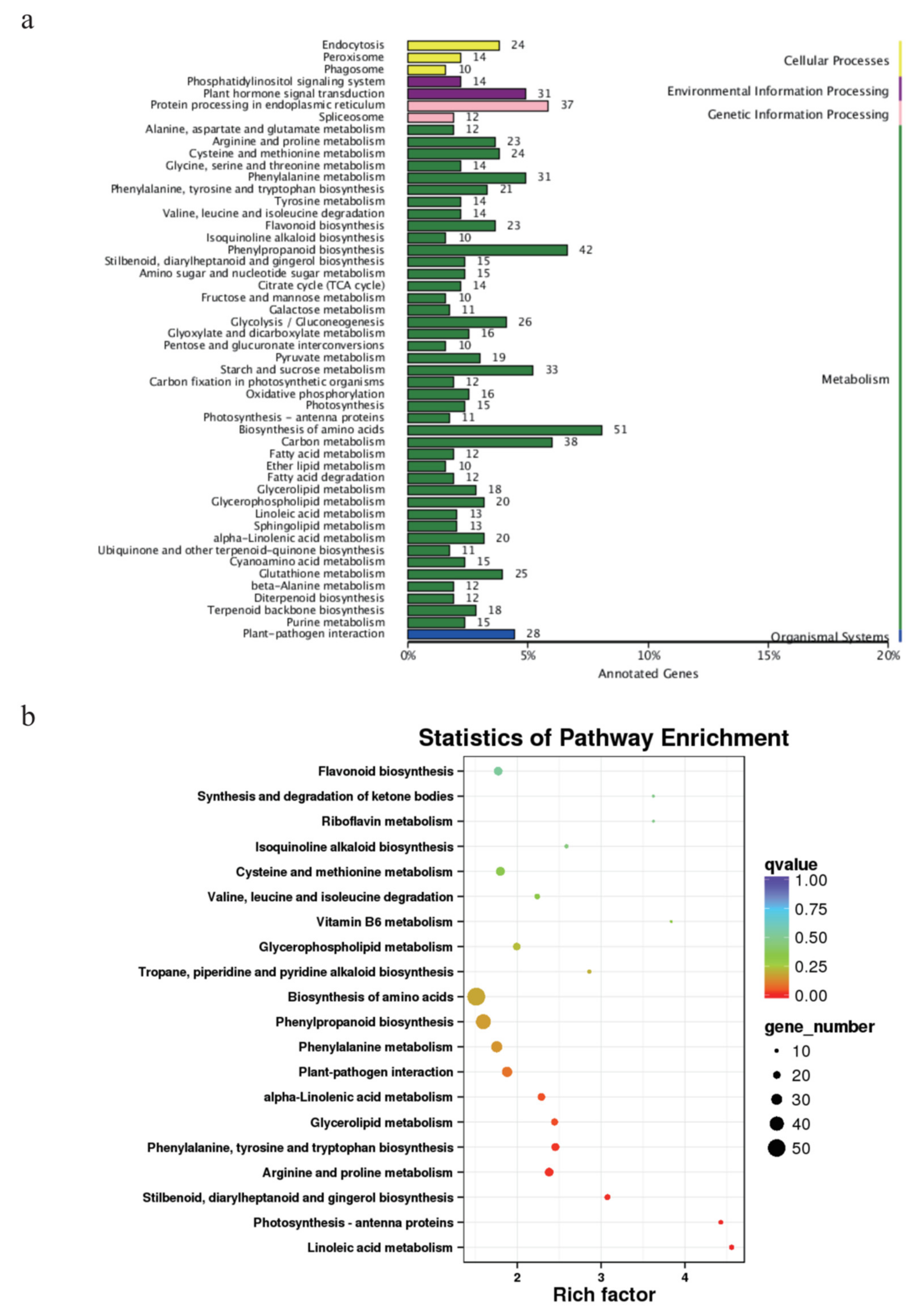
3.4. Growth-Related DEGs in G. biloba Leaves Based on Enrichment and WGCNA Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, Z.; Zheng, S. The missing link in Ginkgo evolution. Nature 2003, 423, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, T.A.; Montoro, P. Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J. Chromatogr. A 2009, 1216, 2002–2032. [Google Scholar] [CrossRef]
- Bastianetto, S.; Zheng, W.H.; Quirion, R. The Ginkgo biloba extract (EGb 761) protects and rescues hippocampal cells against nitric oxide-induced toxicity: Involvement of its flavonoid constituents and protein kinase C. J. Neurochem. 2000, 74, 2268–2277. [Google Scholar] [CrossRef]
- Ihl, R.; Bachinskaya, N.; Korczyn, A.D.; Vakhapova, V.; Tribanek, M.; Hoerr, R.; Napryeyenko, O.; Group, G.S. Efficacy and safety of a once-daily formulation of Ginkgo biloba extract EGb 761 in dementia with neuropsychiatric features: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2011, 26, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, W.; Qin, Y.; Decker, Y.; Wang, X.; Burkart, M.; Schotz, K.; Menger, M.D.; Fassbender, K.; Liu, Y. Long-term treatment with Ginkgo biloba extract EGb 761 improves symptoms and pathology in a transgenic mouse model of Alzheimer’s disease. Brain Behav. Immun. 2015, 46, 121–131. [Google Scholar] [CrossRef]
- Nelissen, H.; Gonzalez, N.; Inze, D. Leaf growth in dicots and monocots: So different yet so alike. Curr. Opin. Plant Biol. 2016, 33, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepworth, J.; Lenhard, M. Regulation of plant lateral-organ growth by modulating cell number and size. Curr. Opin. Plant Biol. 2014, 17, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.; Dengler, N. Leaf Vascular Pattern Formation. Plant Cell 1997, 9, 1121–1135. [Google Scholar] [CrossRef] [Green Version]
- Baute, J.; Herman, D.; Coppens, F.; De Block, J.; Slabbinck, B.; Dell’Acqua, M.; Pe, M.E.; Maere, S.; Nelissen, H.; Inze, D. Combined Large-Scale Phenotyping and Transcriptomics in Maize Reveals a Robust Growth Regulatory Network. Plant Physiol. 2016, 170, 1848–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, M.H.; Sun, J.; Oh, D.H.; Zielinski, R.E.; Clouse, S.D.; Huber, S.C. Enhancing Arabidopsis leaf growth by engineering the BRASSINOSTEROID INSENSITIVE1 receptor kinase. Plant Physiol. 2011, 157, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Liu, J.H.; Zhao, W.S.; Chen, X.J.; Guo, Z.J.; Peng, Y.L. Gibberellin 20-oxidase gene OsGA20ox3 regulates plant stature and disease development in rice. Mol. Plant Microbe Interact. 2013, 26, 227–239. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Gu, Y.; Xu, M.; Wang, J.; Cao, F.; Xu, L.A. Transcriptome analysis of Ginkgo biloba kernels. Front. Plant Sci. 2015, 6, 819. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, L.; Wu, Q.; Men, X.; Yao, L.; Xing, S. Transcriptome profile analysis reveals the ontogenesis of rooted chichi in Ginkgo biloba L. Gene 2018, 669, 8–14. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2, accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, R.; Zhao, Y.; Zhang, H.; Fan, G.; Liu, X.; Zhou, W.; Shi, C.; Wang, J.; Liu, W.; Liang, X.; et al. Draft genome of the living fossil Ginkgo biloba. Gigascience 2016, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.G.; Zhang, G.Q. Single nucleotide polymorphism (SNP) and its application in rice. Yi Chuan 2006, 28, 737–744. [Google Scholar]
- Schmid, K.J.; Sorensen, T.R.; Stracke, R.; Torjek, O.; Altmann, T.; Mitchell-Olds, T.; Weisshaar, B. Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res. 2003, 13, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Huang, S.; Xiao, G.; Zhang, Y.; Yu, J. Abundant RNA editing sites of chloroplast protein-coding genes in Ginkgo biloba and an evolutionary pattern analysis. BMC Plant Biol. 2016, 16, 257. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Choi, D.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003, 36, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Kuijt, S.J.; Greco, R.; Agalou, A.; Shao, J.; t Hoen, C.C.; Overnas, E.; Osnato, M.; Curiale, S.; Meynard, D.; van Gulik, R.; et al. Interaction between the growth-regulating factor and knotted1-like homeobox families of transcription factors. Plant Physiol. 2014, 164, 1952–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschmann, H.; Chan, J.; Sanchez-Pulido, L.; Andrade-Navarro, M.A.; Doonan, J.H.; Lloyd, C.W. Microtubule-associated AIR9 recognizes the cortical division site at preprophase and cell-plate insertion. Curr. Biol. 2006, 16, 1938–1943. [Google Scholar] [CrossRef] [Green Version]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Arlotta, C.; Puglia, G.D.; Genovese, C.; Toscano, V.; Karlova, R.; Beekwilder, J.; De Vos, R.C.H.; Raccuia, S.A. MYB5-like and bHLH influence flavonoid composition in pomegranate. Plant Sci. 2020, 298, 110563. [Google Scholar] [CrossRef] [PubMed]
- Clouse, S.D.; Sasse, J.M. Brassinosteroids: Essential Regulators of Plant Growth and Development. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 427–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.H.; Li, J. BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell 2002, 110, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Geilfus, C.M.; Ober, D.; Eichacker, L.A.; Muhling, K.H.; Zorb, C. Down-regulation of ZmEXPB6 (Zea mays beta-expansin 6) protein is correlated with salt-mediated growth reduction in the leaves of Z. mays L. J. Biol. Chem. 2015, 290, 11235–11245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.T.; Cosgrove, D.J. Altered expression of expansin modulates leaf growth and pedicel abscission in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2000, 97, 9783–9788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Cao, H.; Sun, Y.; Li, X.; Chen, F.; Carles, A.; Li, Y.; Ding, M.; Zhang, C.; Deng, X.; et al. Arabidopsis paired amphipathic helix proteins SNL1 and SNL2 redundantly regulate primary seed dormancy via abscisic acid-ethylene antagonism mediated by histone deacetylation. Plant Cell 2013, 25, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Huarte, H.R.; Puglia, G.D.; Prjibelski, A.D.; Raccuia, S.A. Seed Transcriptome Annotation Reveals Enhanced Expression of Genes Related to ROS Homeostasis and Ethylene Metabolism at Alternating Temperatures in Wild Cardoon. Plants 2020, 9, 1225. [Google Scholar] [CrossRef]
- Puglia, G.D.; Prjibelski, A.D.; Vitale, D.; Bushmanova, E.; Schmid, K.J.; Raccuia, S.A. Hybrid transcriptome sequencing approach improved assembly and gene annotation in Cynara cardunculus (L.). BMC Genom. 2020, 21, 317. [Google Scholar] [CrossRef]
- Smita, S.; Katiyar, A.; Chinnusamy, V.; Pandey, D.M.; Bansal, K.C. Transcriptional Regulatory Network Analysis of MYB Transcription Factor Family Genes in Rice. Front. Plant Sci. 2015, 6, 1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Clean Reads | Mapped Reads | GC (%) | N (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| T01 | 40,843,502 | 32,241,189 (78.94%) | 45.83 | 0.01 | 95.68 | 90.09 |
| T02 | 45,536,702 | 36,635,846 (80.45%) | 45.36 | 0.01 | 95.27 | 89.37 |
| T03 | 43,261,360 | 34,735,785 (80.29%) | 46.00 | 0.01 | 95.61 | 89.94 |
| T04 | 41,269,614 | 33,568,286 (81.34%) | 45.33 | 0.01 | 95.52 | 89.77 |
| T05 | 48,103,982 | 39,385,780 (81.88%) | 45.92 | 0.01 | 95.63 | 89.96 |
| T06 | 46,977,092 | 38,531,630 (82.02%) | 44.70 | 0.01 | 95.21 | 89.27 |
| T07 | 54,312,460 | 44,998,707 (82.85%) | 44.46 | 0.01 | 95.34 | 89.46 |
| T08 | 55,105,380 | 45,855,776 (83.21%) | 44.57 | 0.01 | 95.58 | 89.92 |
| T09 | 58,075,760 | 48,387,657 (83.32%) | 44.57 | 0.01 | 95.68 | 90.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Han, X.; Ruan, J.; Xu, L.; He, B. Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network. Forests 2021, 12, 315. https://doi.org/10.3390/f12030315
Liu H, Han X, Ruan J, Xu L, He B. Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network. Forests. 2021; 12(3):315. https://doi.org/10.3390/f12030315
Chicago/Turabian StyleLiu, Hailin, Xin Han, Jue Ruan, Lian Xu, and Bing He. 2021. "Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network" Forests 12, no. 3: 315. https://doi.org/10.3390/f12030315
APA StyleLiu, H., Han, X., Ruan, J., Xu, L., & He, B. (2021). Transcriptome Analysis of Ginkgo biloba L. Leaves across Late Developmental Stages Based on RNA-Seq and Co-Expression Network. Forests, 12(3), 315. https://doi.org/10.3390/f12030315