
Comparative Analysis of the Complete Chloroplast Genomes of Four Chestnut Species (Castanea)



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, DNA Extraction, and Sequences Sources
2.2. Illumina Sequencing, Assembly, and Annotation
2.3. Repeat Analysis
2.4. Hypervariable Hotspot Identification
2.5. Analysis of DNA Barcodes
2.6. Phylogenetic Analyses
3. Results
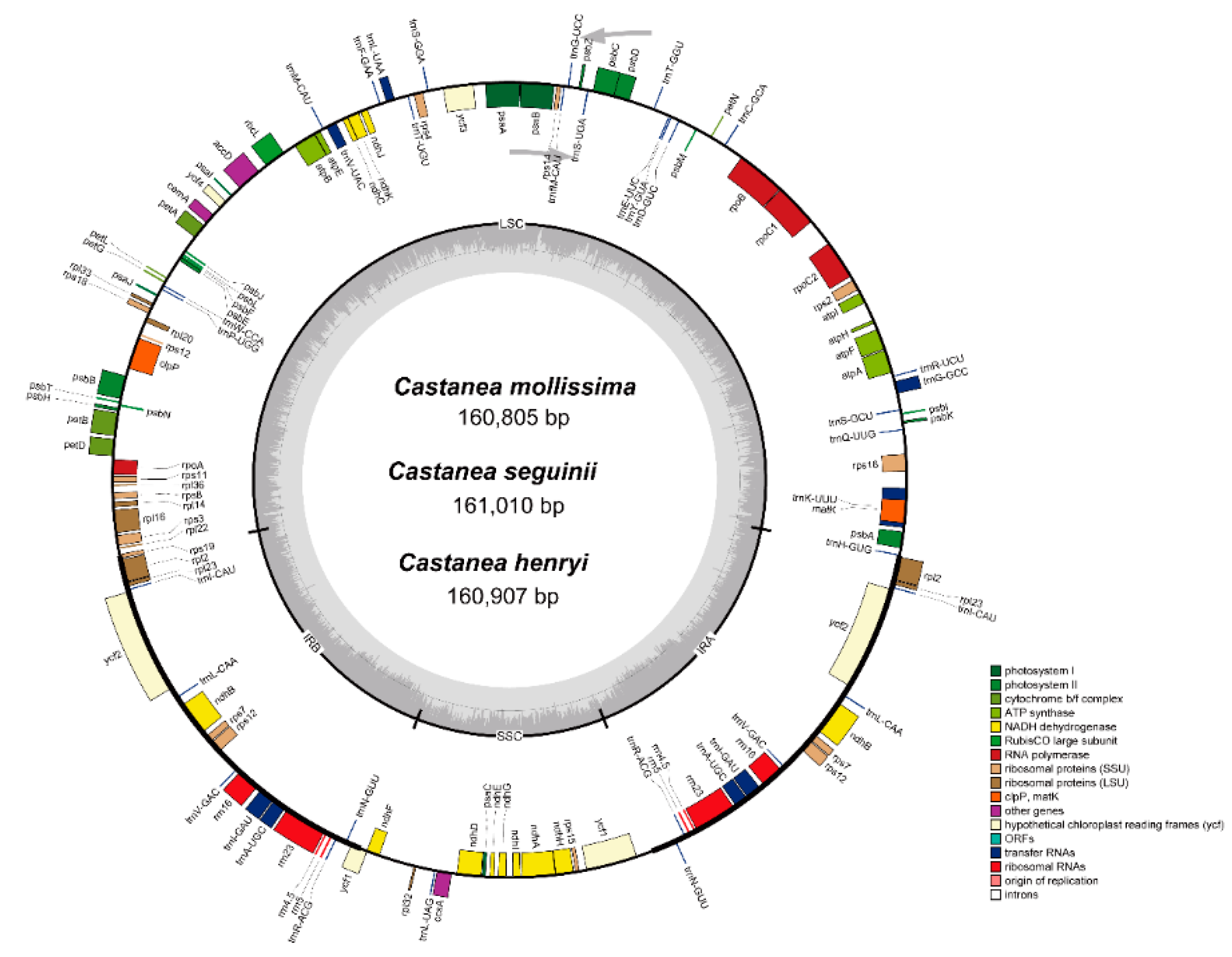
3.1. Characterization and Annotation of the Castanea cp Genomes
3.2. Repeat and Simple Sequence Repeats Analyses
3.3. Selective Pressures in the Evolution of Castanea
3.4. Genome Sequence Divergence
3.5. Phylogenetic Analyses
3.6. Structural and Sequence Comparisons of cp Genomes in Castanea
4. Discussion
4.1. Structural and Sequence Comparisons of cp Genomes in Castanea
4.2. Phylogenetic Analysis
4.3. DNA Barcode Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dane, F.; Lang, P.; Huang, H.; Fu, Y. Intercontinental genetic divergence of Castanea species in eastern Asia and eastern North America. Heredity 2003, 91, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Liu, Y.; Zhang, Q.; Nie, X. Hybrid de novo genome assembly of Chinese chestnut (Castanea mollissima). GigaScience 2019, 8, giz112. [Google Scholar] [CrossRef]
- Barakat, A.; DiLoreto, D.S.; Zhang, Y.; Smith, C.; Baier, K.; Powell, W.A.; Wheeler, N.; Sederoff, R.; Carlson, J.E. Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H. Review of current research of the world Castanea species and importance of germplasm conservation of China native Castanea species. J. Wuhan Bot. Res. 1998, 16, 171–176. [Google Scholar]
- Liu, G.; Fang, J. Spatial patterns of chestnut (Castanea millissima) and its species geographical distribution in China. Acta Ecol. Sin. 2001, 21, 164–170. [Google Scholar]
- Rutter, P.A.; Miller, G.; Payne, J.A. Chestnuts (Castanea). Genet. Resour. Temp. Fruit Nut. Crop. 1991, 290, 761–790. [Google Scholar] [CrossRef]
- Conedera, M.; Krebs, P.; Tinner, W.; Pradella, M.; Torriani, D. The cultivation of Castanea sativa (Mill.) in Europe, from its origin to its diffusion on a continental scale. Veg. Hist. Archaeobot. 2004, 13, 161–179. [Google Scholar] [CrossRef] [Green Version]
- Graves, A.H. Relative blight resistance in species and hybrids of Castanea. Phytopathology 1950, 40, 1125–1131. [Google Scholar]
- Moore, J.N.; Ballington, J.R. Genetic resources of temperate fruit and nut crops. Sci. Hortic. Amst. 1990, 20, 974. [Google Scholar]
- Jacobs, D.F.; Dalgleish, H.J.; Nelson, C.D. A conceptual framework for restoration of threatened plants: The effective model of American chestnut (Castanea dentata) reintroduction. New Phytol. 2013, 197, 111–140. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Dane, F.; Norton, J.D. Allozyme diversity in Chinese, Seguin and American chestnut (Castanea spp.). Theor. Appl. Genet. 1994, 88, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Casasoli, M.; Mattioni, C.; Cherubini, M.; Villani, F. A genetic linkage map of European chestnut (Castanea sativa Mill.) based on RAPD, ISSR and isozyme markers. Theo. Appl. Genet. 2001, 102, 1190–1199. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Liu, Y.; Yuan, Q.; Sun, J.; Guo, L. Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genom. 2021, 22, 103. [Google Scholar]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 1996, 76, 273–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.; Xu, C.; Cheng, T.; Lin, K.; Zhou, S. Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 2013, 5, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef]
- Ding, S.; Dong, X.; Yang, J.; Guo, C.; Cao, B.; Guo, Y.; Hu, G. Complete chloroplast genome of Clethra fargesii Franch, an original sympetalous plant from central China: Comparative analysis, adaptive evolution, and phylogenetic relationships. Forests 2021, 12, 441. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [Green Version]
- Drew, B.T.; Ruhfel, B.R.; Smith, S.A.; Moore, M.J.; Briggs, B.G.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E. Another look at the root of the angiosperms reveals a familiar tale. Syst. Biol. 2014, 63, 368–382. [Google Scholar] [CrossRef] [Green Version]
- Souza, U.J.B.D.; Vitorino, L.C.; Bessa, L.A.; Silva, F.G. The complete plastid genome of Artocarpus camansi: A high degree of conservation of the plastome structure in the family Moraceae. Forests 2020, 11, 1179. [Google Scholar] [CrossRef]
- Alexander, L.; Woeste, K.E. Pyrosequencing of the northern red oak (Quercus rubra L.) chloroplast genome reveals high quality polymorphisms for population management. Tree Genet. Genomes 2014, 10, 803–812. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, L.A.; Liu, J.; Chen, L.; Wu, W.; Hu, W.; Zhang, K.; Kim, S.-C.; Yang, T.J. The complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [Green Version]
- Kane, N.; Sveinsson, S.; Dempewolf, H.; Yang, J.Y.; Zhang, D.P.; Johannes, M.; Engels, M.; Cronk, Q. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 2012, 99, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2017, 7, 1955. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Zhu, Q.; Liu, B. Phylogenetic and biological significance of evolutionary elements from metazoan mitochondrial genomes. PLoS ONE 2014, 9, e84330. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Saski, C.; Lee, S.B.; Hansen, A.K.; Daniell, H. Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2011, 28, 835–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaBonte, N.R.; Zhao, P.; Woeste, K. Signatures of selection in the genomes of Chinese chestnut (Castanea mollissima Blume): The roots of nut tree domestication. Front. Plant Sci. 2018, 9, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Tang, X.; Tu, B.; Xie, P. RAPD analysis of three Chinese endemic Castanea species. J. Fruit Sci. 2004, 3, 275–277. [Google Scholar]
- Gupta, P.K.; Roy, J.K.; Prasad, M. Single nucleotide polymorphisms: A new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants. Curr. Sci. 2001, 80, 524–535. [Google Scholar]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.C.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Dodsworth, S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015, 20, 525–527. [Google Scholar] [CrossRef]
- Pakull, B.; Mader, M.; Kersten, B.; Ekué, M.R.M.; Dipelet, U.G.B.; Paulini, M.; Bouda, H.N.; Degen, B. Development of nuclear, chloroplast and mitochondrial SNP markers for Khaya sp. Conserv. Genet. Resour. 2016, 8, 283–297. [Google Scholar] [CrossRef]
- Dane, F.; Wang, Z.; Goertzen, L. Analysis of the complete chloroplast genome of Castanea pumila var. pumila, the Allegheny chinkapin. Tree Genet. Genomes 2015, 11, 14. [Google Scholar] [CrossRef]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping app. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef]
- Yang, Z. PAML4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.B.; Hao, M.D.; Yang, C.Q.; Shi, Z.Y.; Yu, D. BarcodingR: An integrated rpackage for species identification using DNA barcodes. Methods Ecol. Evol. 2017, 8, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Huang, W.; Lan, Y.; Cao, Q.; Su, S.; Zhou, Z.; Wang, J.; Liu, J.; Hu, G. The complete chloroplast genome sequence of the wild chinese chestnut (Castanea mollissima). Conserv. Genet. Resour. 2018, 10, 291–294. [Google Scholar] [CrossRef]
- Zhu, C.; Shi, F.; Wang, M.; Zhao, Y.; Chen, Y.; Geng, G. The complete chloroplast genome of a variety of Castanea mollissima ‘Hongli’ (Fagaceae). Mitochondrial DNA B 2019, 4, 993–994. [Google Scholar] [CrossRef] [Green Version]
- Zulfiqar, S.; Gao, X.X.; Sun, Y.W.; Hu, G.J.; Zhao, P. The complete chloroplast genome sequence of Seguin chestnut (Castanea seguinii). Mitochondrial DNA B 2019, 4, 342–343. [Google Scholar] [CrossRef]
- Gao, X.X.; Yan, F.; Liu, M.; Zulfiqar, S.; Zhao, P. The complete chloroplast genome sequence of an endemic species Pearl chestnut (castanea henryi). Mitochondrial DNA B 2019, 4, 551–552. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1991, 14, 817–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Angiosperm Phylogeny Group (APG IV). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Ping, L.; Dane, F.; Kubisiak, T.L.; Huang, H. Molecular evidence for an Asian origin and a unique westward migration of species in the genus Castanea via Europe to North America. Mol. Phylogenet. Evol. 2007, 43, 49–59. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Feature | Castanea mollissima1 | Castanea mollissima2 | Castanea mollissima3 | Castanea seguinii1 | Castanea seguinii2 | Castanea henryi1 | Castanea henryi2 | Castanea pumila |
|---|---|---|---|---|---|---|---|---|
| Size (bp) | 160,877 | 160,805 | 160,799 | 160,869 | 161,010 | 160,907 | 160,807 | 160,603 |
| Coding (bp) | 107,291 | 112,516 | 112,340 | 112,299 | 109,149 | 112,525 | 112,720 | 110,191 |
| Noncoding (bp) | 53,586 | 48,289 | 48,459 | 48,570 | 51,861 | 48,382 | 48,087 | 50,412 |
| LSC length (bp) | 90,440 | 90,435 | 90,432 | 90,497 | 90,560 | 90,531 | 90,394 | 90,249 |
| SSC length (bp) | 19,049 | 19,000 | 18,997 | 18,970 | 19,048 | 18,998 | 18,963 | 18,976 |
| IR length (bp) | 51,388 | 51,370 | 51,370 | 51,402 | 51,402 | 51,378 | 51,450 | 51,378 |
| Coding (bp) | 107,291 | 101,832 | 103,345 | 103,249 | 109,511 | 101,843 | 101,104 | 102,650 |
| Noncoding (bp) | 53,586 | 58,973 | 57,454 | 57,620 | 51,499 | 59,064 | 59,703 | 57,953 |
| Number of genes | 130 | 130 | 130 | 130 | 130 | 130 | 130 | 132 |
| Protein-coding genes | 82 | 82 | 83 | 83 | 83 | 82 | 83 | 84 |
| tRNA genes | 37 | 37 | 37 | 37 | 37 | 37 | 37 | 37 |
| rRNA genes | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| GC content (%) | 36.80% | 36.80% | 36.80% | 36.80% | 36.70% | 36.70% | 36.80% | 36.80% |
| Sequencing Platform | Hiseq-PE150 | Hiseq-PE150 | The bridging shotgun | / | Hiseq 4000 | Hiseq 2500 | / | / |
| Raw reads | 24,367,944 | 24,367,945 | / | / | 24,367,947 | 24,367,946 | / | / |
| Raw Base(G) | 11 | 6.6 | / | / | 8.3 | 9 | / | / |
| NCBI Accession | / | / | HQ336406 | NC_033881 | MH998383 | MH998384 | KY951992 | KM360048 |
| Category of Genes | Gene Group | Gene Name | ||||
|---|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA genes | 23S | 16S | 5S | 4.5S | |
| Transfer RNA genes | trnK-UUU | trnI-GAU | trnI-GAU | trnA-UGC | trnG-GCC | |
| trnL-UAA | trnS-UGA | trnL-UAG | trnY-GUA | trnC-GCA | ||
| trnL-CAA | trnH-GUG | trnD-GUC | trnfM-CAU | trnW-CCA | ||
| trnP-UGG | trnI-CAU | trnR-ACG | trnI-CAU | trnE-UUC | ||
| trnT-UGU | trnF-GAA | trnQ-UUG | trnR-UCU | trnT-GGU | ||
| trnM-CAU | trnV-GAC | trnN-GUU | trnV-GAC | trnG-UCC | ||
| trnV-UAC | ||||||
| Small subunit of ribosome | rps12 | rps16 | rps2 | rps3 | rps4 | |
| rps7 | rps11 | rps8 | rps18 | rps15 | ||
| rps14 | rps19 | |||||
| Large subunit of ribosome | rpl16 | rpl2 | rpl14 | rpl20 | rpl23 | |
| rpl33 | rpl32 | rpl36 | ||||
| DNA-dependent RNA polymerase | rpoC1 | rpoC2 | rpoA | rpoB | ||
| Genes for photosynthesis | Subunits of NADH-dehydrogenase | ndhA | ndhB | ndhD | ndhH | ndhF |
| ndhK | ndhG | ndhI | ndhJ | ndhC | ||
| ndhE | ||||||
| Subunits of photosystem I | psaA | psaB | psaC | psaI | psaJ | |
| ycf1 | ycf2 | ycf4 | ycf3 | |||
| Subunits of photosystem II | psbB | psbC | psbA | psbD | psbE | |
| psbH | psbZ | psbK | psbN | psbJ | ||
| psbF | psbL | psbI | psbT | psbM | ||
| Subunits of cytochrome b/f complex | petB | petD | petA | petG | petL | |
| petN | ||||||
| Subunits of ATP synthase | atpA | atpB | atpF | atpI | atpE | |
| atpH | ||||||
| Subunits of rubisco | rbcL | |||||
| Other genes | Maturase | matK | ||||
| Protease | clpP | |||||
| Envelope membrane protein | cemA | |||||
| Subunit of Acetyl-CoA-carboxylase | accD | |||||
| C-type cytochrome synthesis gene | ccsA | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Gao, X.; Woeste, K.; Zhao, P.; Zhang, S. Comparative Analysis of the Complete Chloroplast Genomes of Four Chestnut Species (Castanea). Forests 2021, 12, 861. https://doi.org/10.3390/f12070861
Zhou H, Gao X, Woeste K, Zhao P, Zhang S. Comparative Analysis of the Complete Chloroplast Genomes of Four Chestnut Species (Castanea). Forests. 2021; 12(7):861. https://doi.org/10.3390/f12070861
Chicago/Turabian StyleZhou, Huijuan, Xiaoxiao Gao, Keith Woeste, Peng Zhao, and Shuoxin Zhang. 2021. "Comparative Analysis of the Complete Chloroplast Genomes of Four Chestnut Species (Castanea)" Forests 12, no. 7: 861. https://doi.org/10.3390/f12070861
APA StyleZhou, H., Gao, X., Woeste, K., Zhao, P., & Zhang, S. (2021). Comparative Analysis of the Complete Chloroplast Genomes of Four Chestnut Species (Castanea). Forests, 12(7), 861. https://doi.org/10.3390/f12070861