
Assessing the Genetic Identity of Tuscan Sweet Chestnut (Castanea sativa Mill.)



Abstract
:1. Introduction
2. Materials and Methods
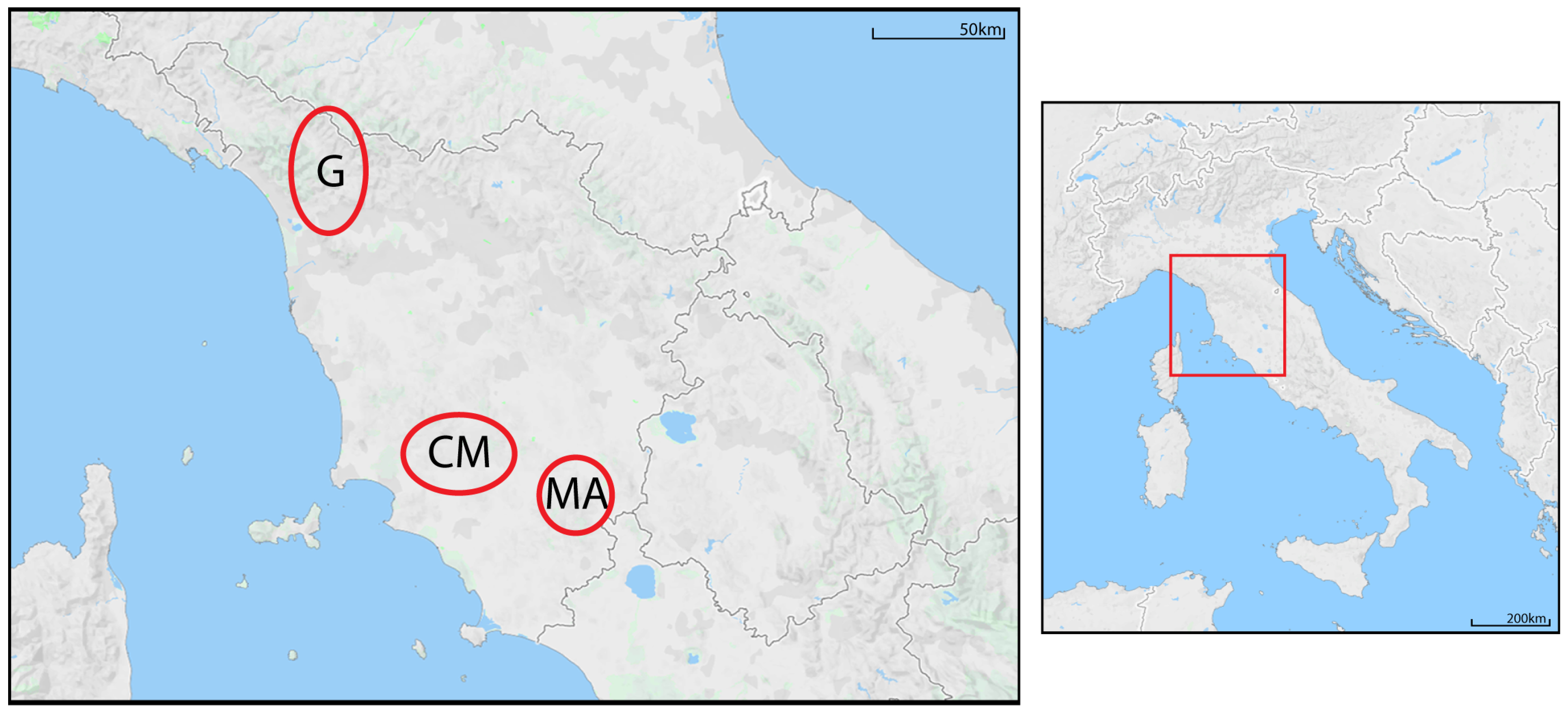
2.1. Study Area
2.2. Genetic Analysis
2.3. Demography
3. Results
3.1. Genetic Diversity
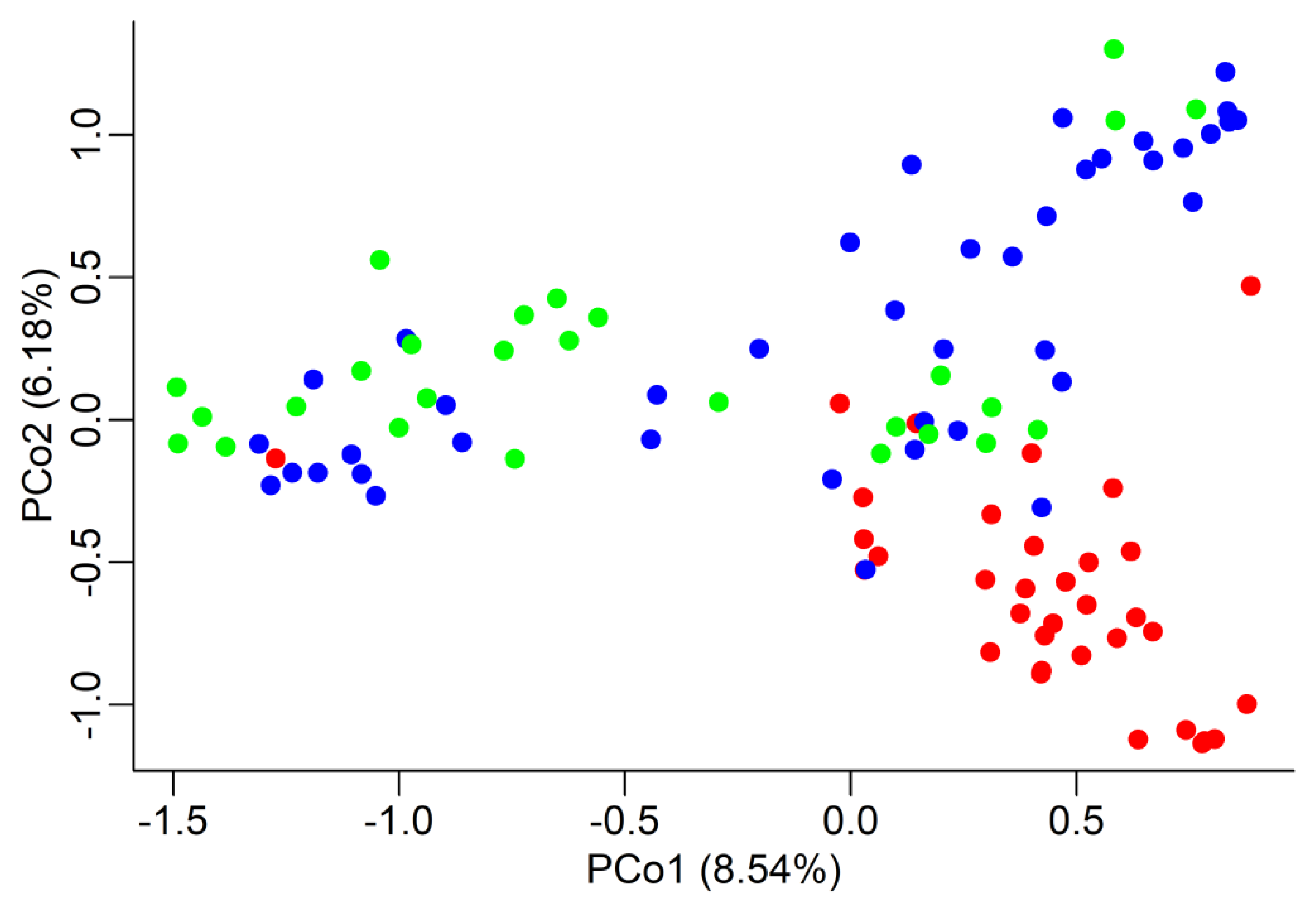
3.2. Genetic Differentiation and Distance
3.3. Structure Analysis
3.4. Demographic History
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neri, L.; Dimitri, G.; Sacchetti, G. Chemical composition and antioxidant activity of cured chestnuts from tree sweet chestnut (Castanea sativa Mill.) ecotypes from Italy. J. Food Compos. Anal. 2010, 23, 23–29. [Google Scholar] [CrossRef]
- Jaynes, R.A. Chestnut chromosomes. For. Sci. 1962, 13, 372–377. [Google Scholar]
- Alessandri, S.; Cabrer, A.M.R.; Martìn, M.A.; Mattioni, C.; Pereira-Lorenzo, S.; Dondini, L. Genetic characterization of Italian and Spanish wild and domesticated chestnut tress. Sci. Hortic. 2022, 295, 110882. [Google Scholar] [CrossRef]
- Conedera, M.; Krebs, P.; Tinner, W.; Pradella, M.; Torriani, D. The cultivation of Castanea sativa (Mill.) in Europe, from its origin to its diffusion on a continental scale. Veg. Hist. Archaeobot. 2004, 13, 161–179. [Google Scholar] [CrossRef] [Green Version]
- Zohary, D.; Hopf, M. Domestication of Plants in the Old World; Claredon Press: Oxford, UK, 1988. [Google Scholar]
- Casoli, M.; Mattioni, C.; Cherubini, M.; Villani, F. A genetic linkage map of European chestnut (Castanea sativa Mill.) based on RAPD, ISSR and isozyme markers. Theor. Appl. Genet. 2001, 102, 1190–1199. [Google Scholar] [CrossRef]
- Barreneche, T.; Casasoli, M.; Russell, K.; Akkak, A.; Meddour, H.; Plomion, C.; Villani, F.; Kremer, A. Comparative mapping between Quercus and Castanea using simple-sequence repeats (SSRs). Theor. Appl. Genet. 2004, 108, 558–566. [Google Scholar] [CrossRef]
- Staton, M.; Zhebentyayeva, T.; Olukolu, B.; Fang, G.C.; Nelson, D.; Carlson, J.E.; Abbott, A.G. Substantial genome synteny preservation among woody angiosperm species: Comparative genomics of Chinese chestnut (Castanea mollissima) and plant reference genomes. BMC Genom. 2015, 16, 744. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.; Antorrena, G.; Freire, M.S.; Pizzi, A.; Gonzàles-Alvàrez, J. Environmentally friendly wood adhesives based on chestnut (Castanea sativa) shell tannins. Eur. J. Wood Wood Prod. 2017, 75, 89–100. [Google Scholar] [CrossRef]
- Mattioni, C.; Martin, M.A.; Pollegioni, P.; Cherubini, M.; Villani, F. Microsatellite markers reveal a strong geographical structure in European populations of Castanea sativa (Fagaceae): Evidence for multiple glacial refugia. Am. J. Bot. 2013, 100, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Huntley, B.; Birks, H.J.B. An Atlas of Past and Present Pollen Maps for Europe: 0–13,000 Years Ago; Cambridge University Press: Cambridge, UK, 1983; p. 688. [Google Scholar]
- Fineschi, S.; Malvolti, M.E.; Morganti, M.; Vendramin, G.G. Allozyme variation within and among cultivated varieties of sweet chestnut (Castanea sativa). Can. J. For. Res. 1994, 24, 1160–1165. [Google Scholar] [CrossRef]
- Villani, F.; Pigliucci, M.; Cherubini, M. Evolution of Castanea sativa Mill. in Turkey and Europe. Genet. Res. 1994, 63, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Villani, F.; Sansotta, A.; Cherubini, M.; Cesaroni, D.; Sbordoni, V. Genetic structure of natural population of Castanea sativa in Turkey: Evidence of a hybrid zone. J. Evol. Biol. 1999, 12, 233–244. [Google Scholar] [CrossRef]
- Galderisi, U.; Cipollaro, M.; Bernardo, G.; Masi, L.; Galano, G.; Cascino, A. Molecular typing of Italian sweet chestnut cultivars by random amplified polymorphic DNA analysis. J. Hortic. Sci. Biotech. 1998, 73, 259–263. [Google Scholar] [CrossRef]
- Boccacci, P.; Akkak, A.; Marinoni, D.; Bounous, G.; Botta, R. Typing european chestnut (Castanea sativa Mill.) cultivars using oak simple sequence repeat markers. HortScience 2004, 39, 1212–1216. [Google Scholar] [CrossRef] [Green Version]
- Botta, R.; Akkak, A.; Guaraldo, P.; Bounous, G. Genetic characterization and nut quality of chestnut cultivars from Piemonte (Italy). Acta Hortic. 2005, 693, 395–401. [Google Scholar] [CrossRef]
- Mattioni, C.; Cherubini, M.; Micheli, E.; Villani, F.; Bucci, G. Role of domestication in shaping Castanea sativa genetic variation in Europe. Tree Genet. Genomes 2008, 4, 563–574. [Google Scholar] [CrossRef]
- Martin, M.A.; Mattioni, C.; Cherubini, M.; Taurchini, D.; Villani, F. Genetic characterization of traditional chestnut varieties in Italy using microsatellites (simple sequence repeats) markers. Ann. Appl. Biol. 2010, 157, 37–44. [Google Scholar] [CrossRef]
- Alessandri, S.; Krznar, M.; Ajolfi, D.; Ramos Cabrer, A.M.; Pereira-Lorenzo, S.; Dondini, L. Genetic Diversity of Castanea sativa Mill. Accessions from the Tuscan-Emilian Apennines and Emilia Romagna Region (Italy). Agronomy 2020, 10, 1319. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Marinoni, D.; Akkak, A.; Bounous, G.; Edwards, K.J.; Botta, R. Development and characterization of microsatellite markers in Castanea sativa (Mill.). Mol. Breed. 2003, 11, 127–136. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Buck, E.J.; Hadonou, M.; James, C.J.; Blakesley, D.; Russell, K. Isolation and characterization of polymorphic microsatellites in European chestnut (Castanea sativa Mill.). Mol. Ecol. Notes 2003, 3, 239–241. [Google Scholar] [CrossRef]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX 4.02, Logiciel Sous WindowsTM Pour la Génétique des Populations; Laboratoire Génome, Population Interaction, CNRS; Université de Montpellier II: Montpellier, France, 1996. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Dent, E.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Santos, F.; Beaumont, M.A. Inferring population history with DIY ABC: A user-friendly approach to approximate Bayesian computation. Bioinformatics 2008, 24, 2713–2719. [Google Scholar] [CrossRef] [Green Version]
- Barthe, S.; Binelli, G.; Hérault, B.; Scotti-Saintagne, C.; Sabatier, D.; Scotti, I. Tropical rainforests that persisted: Inferences from the Quaternary demographic history of eight tree species in the Guiana shield. Mol. Ecol. 2017, 26, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.A.; Zhang, W.; Balding, D.J. Approximate Bayesian computation in population genetics. Genetics 2002, 162, 2025–2035. [Google Scholar] [CrossRef]
- Bowman, J.; Greenhorn, J.E.; Marrotte, R.R.; McKay, M.M.; Morris, K.Y.; Mrentice, M.B.; Wehtje, M. On application of landscape genetics. Conserv. Genet. 2016, 17, 753–760. [Google Scholar] [CrossRef]
- Mattioni, C.; Martin, M.A.; Chiocchini, F.; Cherubini, M.; Gaudet, M.; Pollegioni, P.; Velichkov, I.; Jarman, R.; Chambers, F.M.; Paule, L.; et al. Landscape genetics structure of European sweet chestnut (Castanea sativa Mill): Indications for conservation priorities. Tree Genet. Genomes 2017, 13, 39. [Google Scholar] [CrossRef]
- Poljak, I.; Idžojtić, M.; Šatović, Z.; Ježić, M.; Ćurković-Perica, M.; Simovski, B.; Acevski, J.; Liber, Z. Genetic diversity of the sweet chestnut (Castanea sativa Mill.) in Central Europe and the western part of the Balkan Peninsula and evidence of marron genotype introgression into wild populations. Tree Genet. Genomes 2017, 13, 18. [Google Scholar] [CrossRef]
- Cuestas, M.I.; Mattioni, C.; Martin, L.M.; Vargas-Osuna, E.; Cherubini, M.; Martin, M.A. Functional genetic diversity of chestnut (Castanea sativa Mill.) populations from southern Spain. For. Syst. 2017, 26, eSC06. [Google Scholar] [CrossRef] [Green Version]
- Lusini, I.; Velichkov, I.; Pollegioni, P.; Chiocchini, F.; Hinkov, G.; Zlatanov, T.; Cherubini, M.; Mattioni, C. Estimating the genetic diversity and spatial structure of Bulgarian Castanea sativa populations by SSRs: Implications for conservation. Conser. Genet. 2014, 15, 283–293. [Google Scholar] [CrossRef]
- Fernàndez-Cruz, J.; Fernàndez-Lòpez, J. Genetic structure of wild sweet chestnut (Castanea sativa Mill.) populations in northwest of Spain and their differences with other European stands. Conserv. Genet. 2016, 17, 949–967. [Google Scholar] [CrossRef]
- Martin, M.A.; Mattioni, C.; Molina, J.R.; Alvarez, J.B.; Cherubini, M.; Herrera, M.A.; Villani, F.; Martin, L.M. Landscape genetic structure of chestnut (Castanea sativa Mill.) in Spain. Tree Genet. Genomes 2012, 8, 127–136. [Google Scholar] [CrossRef]
- Hubbell, S.P. The Unified Neutral Theory of Biodiversity and Biogeography; Princeton University Press: Princeton, NJ, USA, 2001. [Google Scholar]
- Exposito-Alonso, M.; Vasseur, F.; Ding, W.; Wang, G.; Burbano, H.A.; Weigel, D. Genomic basis and evolutionary potential for extreme drought adaptation in Arabidopsis thaliana. Nat. Ecol. Evol. 2018, 2, 352–358. [Google Scholar] [CrossRef]
- Di Pierro, E.A.; Mosca, E.; González-Martínez, S.C.; Binelli, G.; Neale, D.B.; La Porta, N. Adaptive variation in natural Alpine populations of Norway spruce (Picea abies [L.] Karst) at regional scale: Landscape features and altitudinal gradient effects. For. Ecol. Manag. 2017, 405, 350–359. [Google Scholar] [CrossRef]
- Yücedağ, C.; Müller, M.; Gailing, O. Morphological and genetic variation in natural populations of Quercus vulcanica and Q. frainetto. Plant Syst. Evol. 2021, 307, 8. [Google Scholar] [CrossRef]
- Castellana, S.; Martin, M.Á.; Solla, A.; Alcaide, F.; Villani, F.; Cherubini, M.; Neale, D.; Mattioni, C. Signatures of local adaptation to climate in natural populations of sweet chestnut (Castanea sativa Mill.) from southern Europe. Ann. For. Sci. 2021, 78, 27. [Google Scholar] [CrossRef]
- Larue, C.; Guichoux, E.; Laurent, B.; Barreneche, T.; Robin, C.; Massot, M.; Delcamp, A.; Petit, R.J. Development of highly validated SNP markers for genetic analyses of chestnut species. Conserv. Genet. Resour. 2021, 13, 383–388. [Google Scholar] [CrossRef]
- Zhivotovsky, L.A.; Feldman, M.W.; Grishechkin, S.A. Biased mutations and microsatellite variation. Mol. Biol. Evol. 1997, 14, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Estoup, A.; Jarne, P.; Cornuet, J.M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 2002, 11, 1591–1604. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Ghirotto, S.; Mona, S.; Benazzo, A.; Paparazzo, F.; Caramelli, D.; Barbujani, G. Inferring genealogical processes from patterns of Bronze-Age and modern DNA variation in Sardinia. Mol. Biol. Evol. 2010, 27, 875–886. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Locus | Primer Sequence (5’–3’) | Ta | |
|---|---|---|---|
| CsCAT1-NED | F: | GAGAATGCCCACTTTTGCA | 50 °C |
| R: | GCTCCCTTATGGTCTCG | ||
| CsCAT3-FAM | F: | CACTATTTTATCATGGACGG | 50 °C |
| R: | CGAATTGAGAGTTCATACTC | ||
| CSCAT6-HEX | F: | AGTGCTCGTGGTCAGTGAG | 50 °C |
| R: | CAACTCTGCATGATAAC | ||
| CsCAT16-HEX | F: | CTCCTTGACTTTGAAGTTGC | 50 °C |
| R: | CTGATCGAGAGTAATAAAG | ||
| CSCAT17-FAM | F: | TTGGCTATACTTGTTCTGCAAG | 58 °C |
| R: | GCCCCATGTTTTCTTCCATGG | ||
| EMCs22-FAM | F: | GTGCCTCTGTATGCATGGTAAGC | 60 °C |
| R: | CCAGGTTTAAGAAAGCAAGCATAAC | ||
| EMCs25-HEX | F: | ATGGGAAAATGGGTAAAGCAGTAA | 58 °C |
| R: | AACCGGAGATAGGATTGAACAGAA | ||
| EMCs38-NED | F: | TTTCCCTATTTCTAGTTTGTGATG | 56 °C |
| R: | ATGGCGTTTGGATGAAC |
| Locus | Population | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Garfagnana (G) | Colline Metallifere (CM) | Monte Amiata (MA) | ||||||||
| He | Ho | FIS | He | Ho | FIS | He | Ho | FIS | PIC | |
| CsCAT1 | 0.601 | 0.735 | −0.210 | 0.587 | 0.525 | 0.118 | 0.744 | 0.739 | 0.029 | 0.6204 |
| CsCAT3 | 0.770 | 0.647 | 0.174 | 0.719 | 0.585 | 0.198 | 0.754 | 0.760 | 0.012 | 0.7321 |
| CsCAT6 | 0.826 | 0.912 | −0.089 | 0.808 | 0.825 | −0.08 | 0.780 | 0.926 | −0.169 | 0.8556 |
| CsCAT16 | 0.715 | 0.824 | −0.137 | 0.595 | 0.463 | 0.233 | 0.690 | 0.741 | −0.055 | 0.6691 |
| CsCAT17 | 0.686 | 0.765 | −0.101 | 0.809 | 0.951 | −0.164 | 0.733 | 0.926 | −0.245 | 0.8028 |
| EMC22 | 0.698 | 0.794 | −0.123 | 0.817 | 0.756 | 0.087 | 0.774 | 0.615 | 0.223 | 0.7874 |
| EMC25 | 0.371 | 0.029 | 0.923 | 0.688 | 0.512 | 0.267 | 0.645 | 0.370 | 0.441 | 0.7339 |
| EMC38 | 0.793 | 0.765 | 0.051 | 0.891 | 0.889 | 0.017 | 0.842 | 0.913 | −0.062 | 0.8841 |
| Mean/overall FIS | 0.682 | 0.684 | 0.013 | 0.740 | 0.688 | 0.082 | 0.745 | 0.749 | 0.015 | |
| Given | Inferred Clusters | N° | ||
|---|---|---|---|---|
| Pop | 1 | 2 | 3 | |
| G: | 0.960 | 0.011 | 0.029 | 34 |
| CM: | 0.000 | 0.730 | 0.270 | 41 |
| MA: | 0.000 | 0.415 | 0.585 | 27 |
| Gene Pool | Scenario | T | N0µ0 | N1µ1 | r0 |
|---|---|---|---|---|---|
| Garfagnana w/o 17 | Contraction | 783 (20.2–7940) | 0.70 (0.034–5.80) | 17.2 (3.24–63.5) | 0.041 |
| Purple cluster | Expansion | 1080 (21.7–9450) | 10.3 (0.40–62.5) | 4.91 (0.44–62.5) | 2.10 |
| Orange cluster | Contraction | 1350 (24.7–9070) | 1.31 (0.06–9.40) | 11.9 (2.24–53.3) | 0.110 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavallini, M.; Lombardo, G.; Binelli, G.; Cantini, C. Assessing the Genetic Identity of Tuscan Sweet Chestnut (Castanea sativa Mill.). Forests 2022, 13, 967. https://doi.org/10.3390/f13070967
Cavallini M, Lombardo G, Binelli G, Cantini C. Assessing the Genetic Identity of Tuscan Sweet Chestnut (Castanea sativa Mill.). Forests. 2022; 13(7):967. https://doi.org/10.3390/f13070967
Chicago/Turabian StyleCavallini, Marta, Gianluca Lombardo, Giorgio Binelli, and Claudio Cantini. 2022. "Assessing the Genetic Identity of Tuscan Sweet Chestnut (Castanea sativa Mill.)" Forests 13, no. 7: 967. https://doi.org/10.3390/f13070967
APA StyleCavallini, M., Lombardo, G., Binelli, G., & Cantini, C. (2022). Assessing the Genetic Identity of Tuscan Sweet Chestnut (Castanea sativa Mill.). Forests, 13(7), 967. https://doi.org/10.3390/f13070967