
Transcriptome Sequencing and Analysis of Genes Related to Disease Resistance in Pinus thunbergii

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. RNA Extraction, Library Construction and Sequencing
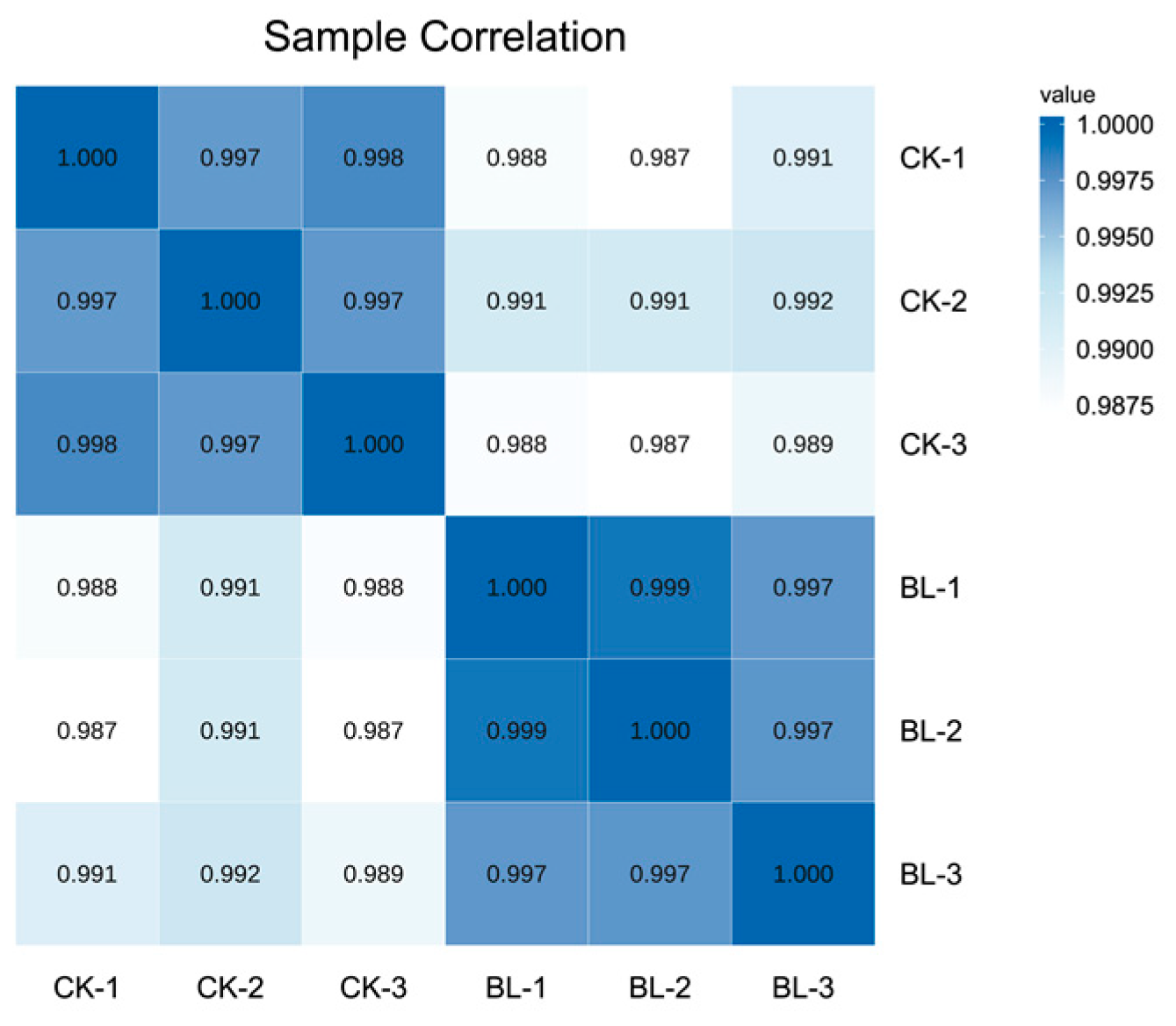
2.3. Differentially Expressed Genes (DEGs) and Relationship Analysis of Samples
2.4. Fluorescence Quantitative PCR Analysis
3. Results and Analysis
3.1. Infection and Identification of PWN in Black Pine Seedlings
3.2. Transcriptome Sequencing and Assembly
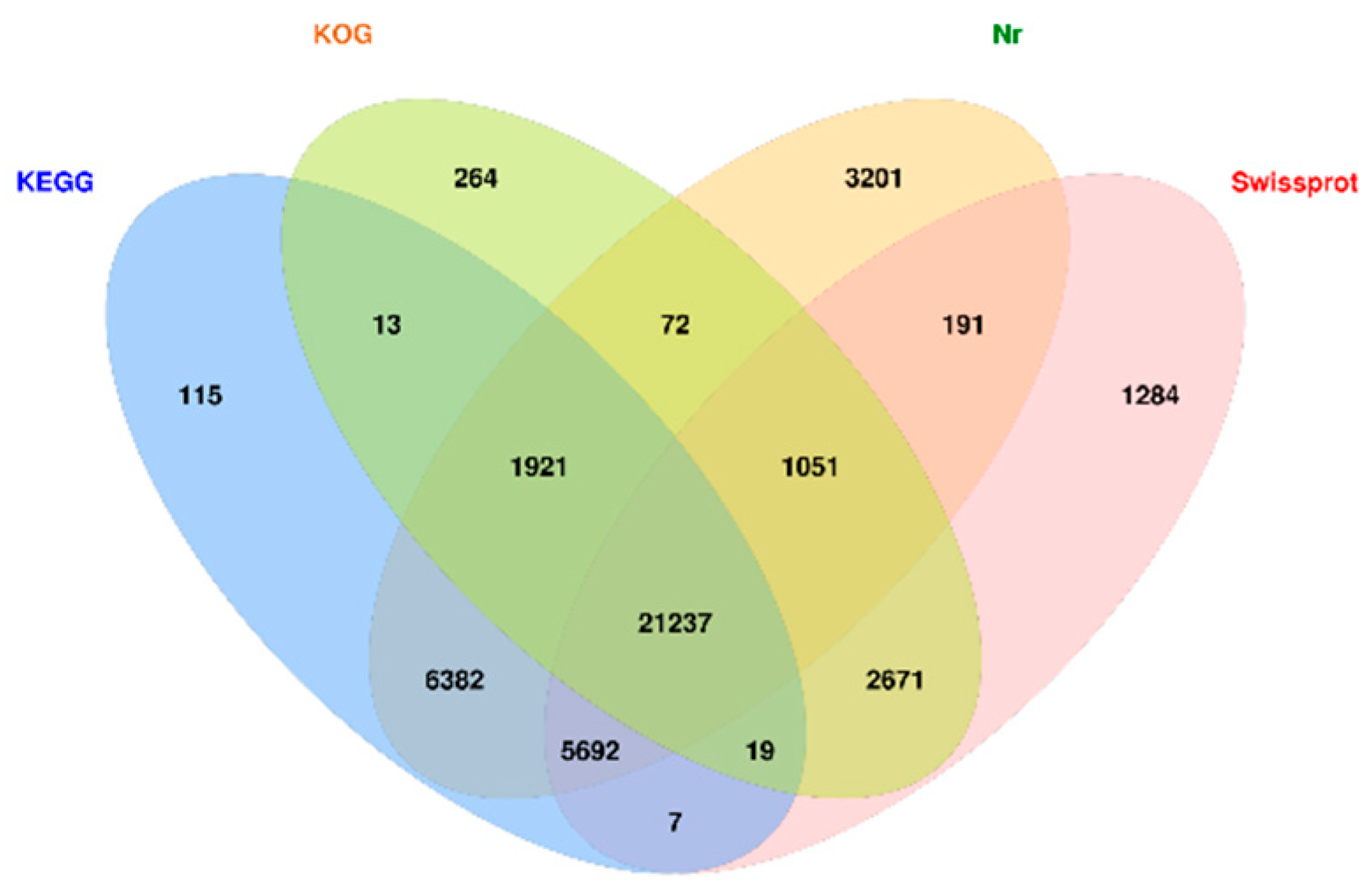
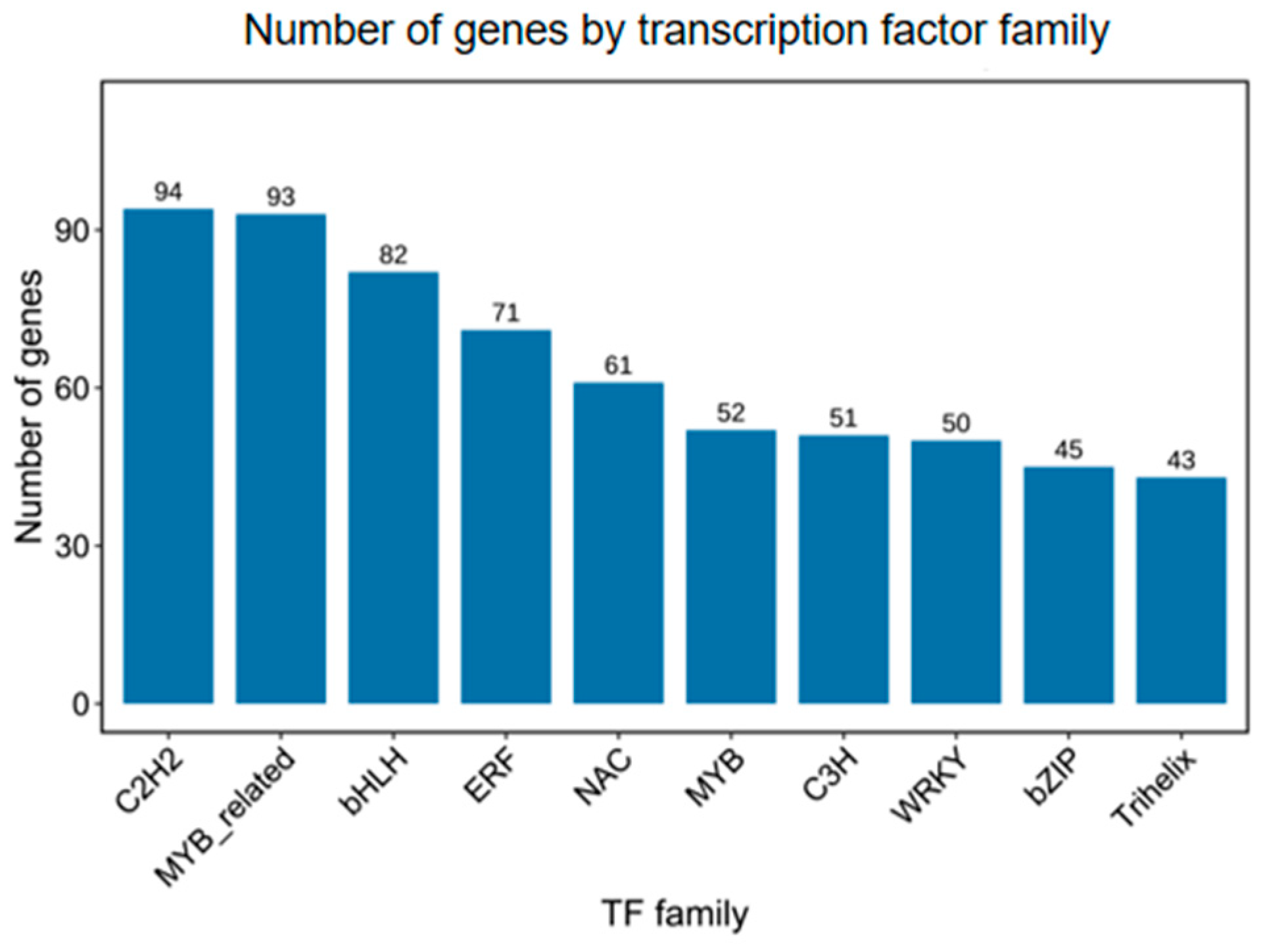
3.3. Basic and Advanced Annotates of Unigenes
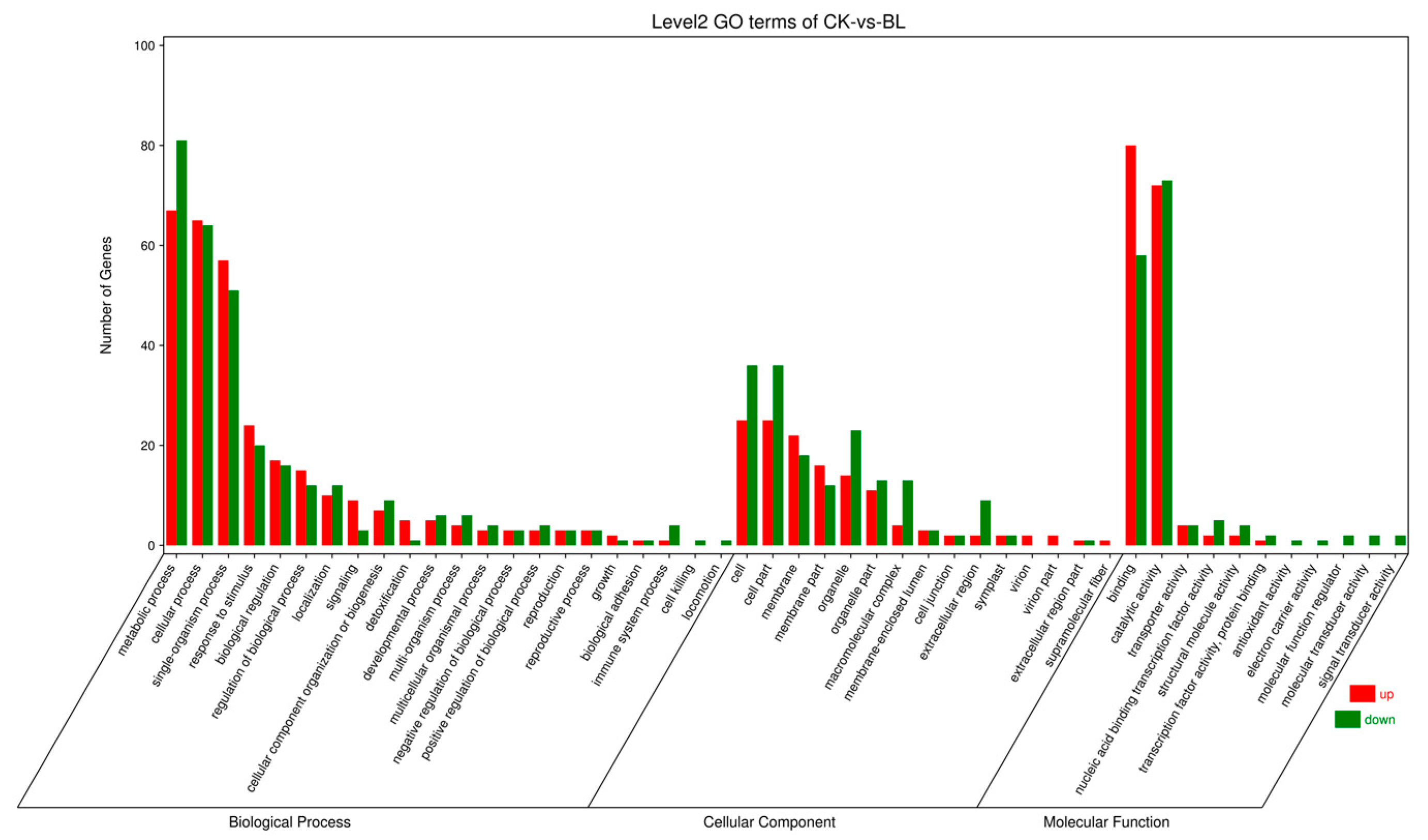
3.4. Differential Gene Analysis
3.5. PCR Analysis Results
3.6. Screening Disease-Resistance-Related DEGs
4. Discussions
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hu, S.Y.; Li, Y.; Yang, X.Y.; Sun, J.S. Preliminary investigation on vascular plant in Yantai coastal shelterbelt provincial natural reserve. Pop. Sci. Technol. 2022, 24, 15–18. [Google Scholar]
- Nickle, W.R. A taxonomic review of the genera of the Aphelenchoidea (fuchs) thorne, 1949 (nematoda: Ty-lenchida). J. Nematol. 1970, 2, 375–392. [Google Scholar]
- Steiner, G.; Buhrer, E.M. Aphelenchoides xylophilus n. sp., a nematode associated with blue-stain and other fungi in timber. J. Agric. Res. 1934, 48, 949–951. [Google Scholar]
- Li, H.; Xia, X.; He, X.; Li, S.; Dai, L.; Ye, J.; Hao, D. Comparative transcriptome analysis reveals molecular insights in overwintering Monochamus alternatus (Coleoptera: Cerambycidae). J. Insect. Sci. 2022, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.M.; Zhang, D.H.; Zhang, G.X.; Luo, J.P.; Zhou, G.X. Adults of pine longhorn beetle carry the number of pine wood nematodes. J. Northeast For. Univ. 2000, 5, 99–101. [Google Scholar]
- Cheng, H.R.; Lin, M.; Li, W.; Fang, Z. The occurrence of a pine wilting disease caused by nematode found in Nanjing. For. Pest. Dis. 1983, 4, 5. [Google Scholar]
- Ruan, X.S. Occurrence and control of pine wood nematode in Taiwan. Plant Quar. 1996, 2, 86–87. [Google Scholar]
- Yi, C.K.; Byun, B.H.; Park, J.D.; Yang, S.I.; Chang, K.H. First finding of the pine wood nematode, Bursaphelenchus xylophilus (Steiner & Buhrer) Nickle and its insect vector in Korea. Res. Rep. For. Res. Inst. Seoul 1998, 38, 141–149. [Google Scholar]
- Wang, X.Y.; Wu, X.Q.; Wen, T.Y.; Feng, Y.Q.; Zhang, Y. Terpene production varies in Pinus thunbergii Parl. with different levels of resistance, with potential effects on pinewood nematode behavior. Forests 2022, 13, 1140. [Google Scholar] [CrossRef]
- Kuroda, K. Mechanism of cavitation development in the pine wilt disease. For. Pathol. 1991, 21, 82–89. [Google Scholar] [CrossRef]
- Xie, W.F.; Liang, G.H.; Huang, A.Z.; Zhang, F.P.; Guo, W.S. Comparative study on the mRNA expression of Pinus massoniana infected by Bursaphelenchus xylophilus. J. For. Res. 2020, 31, 75–86. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; Langer, S.; Carrasquinho, I.; Bergström, E.; Larson, T.; Thomas-Oates, J.; António, C. Pinus pinaster early hormonal defence responses to pinewood nematode (Bursaphelenchus xylophilus) infection. Metabolites 2021, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Meireles, B.; Fortes, A.M. Comparative transcriptomic response of two pinus species to infection with the pine wood nematode Bursaphelenchus xylophilus. Forests 2020, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Han, H.; Koh, Y.H.; Kim, I.S.; Lee, S.W.; Shim, D. Comparative transcriptome analysis of Pinus densiflora following inoculation with pathogenic (Bursaphelenchus xylophilus) or non-pathogenic nematodes (B. thailandae). Sci. Rep. 2019, 219, 12180. [Google Scholar] [CrossRef] [Green Version]
- Li, C.F.; Xu, Y.X.; Ma, J.Q.; Jin, J.Q.; Huang, D.J.; Yao, M.Z.; Ma, C.L.; Chen, L. Biochemical and transcriptomic analyses reveal different metabolite biosynthesis profiles among three color and developmental stages in ‘Anji Baicha’ (Camellia sinensis). BMC Plant Biol. 2016, 16, 195. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.P.; Able, A.J.; Able, J.A. Integrated analysis of small RNA, transcriptome, and degradome sequencing reveals the water-deficit and heat stress response network in durum wheat. Int. J. Mol. Sci. 2020, 21, 6017. [Google Scholar] [CrossRef]
- Zhou, H.; Shen, P.Y.; Ye, C.Q. Improvement of nematode separator and isolation characteristics of pine wood nematodes. Plant Quar. 2002, 16, 3. [Google Scholar]
- Zhang, R.Z. Effects of inoculation method and inoculation source concentration on the occurrence of wilt and withering of pine wood nematodes. Taiwan For. Sci. 1999, 14, 409–417. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNAseq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Qi, Z.; Du, Y.; Yu, J.; Zhang, R.; Yu, M.; Cao, H.; Song, T.; Pan, X.; Liang, D.; Liu, Y. Molecular detection and analysis of blast resistance genes in rice main varieties in Jiangsu province, China. Agronomy 2023, 13, 157. [Google Scholar] [CrossRef]
- Maruyama, Y.; Yamoto, N.; Suzuki, Y.; Chiba, Y.; Yamazaki, K.; Sato, T.; Yamaguchi, J. The Arabidopsis transcriptional repressor ERF9 participate in resistance against necrotrophic fungi. Plant Sci. 2013, 213, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Luan, Y.S.; Li, J.B.; Yin, Y.L. Expression of a tomato MYB gene in transgenic tobacco increases resistance to Fusarium oxysporum and Botrytis cinerea. Eur. J. Plant Pathol. 2016, 144, 607–617. [Google Scholar] [CrossRef]
- Xi, D.; Yin, T.; Han, P.; Yang, X.; Zhang, M.; Du, C.; Zhang, H.; Liu, X. Genome-wide identification of sweet orange WRKY transcription factors and analysis of their expression in response to infection by Penicillium digitatum. Curr. Issues Mol. Biol. 2023, 45, 1250–1271. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.S.; Wang, Y.F.; Wang, R.; Liu, Y.R.; Gu, L.F.; Zhou, P.; Yin, W.X.; Luo, C.X. Functional study on bZIP transcription factor UvbZIP12 in Ustilaginoidea virens. Acta Phytopathol. Sin. 2022, 52, 555–564. [Google Scholar]
- Sun, Z.X.; Wei, D.Q.; Yang, M.; Lan, X.G. Biosynthesis and function of plant thiamine. Acta. Physiol. plant. 2018, 54, 1791–1796. [Google Scholar]
- Ge, Y.H.; Li, C.Y.; Lu, J.Y.; Zhou, D.S. Effects of thiamine on Trichothecium and Alternaria rots of muskmelon fruit and the possible mechanisms involved. J. Integr. Agric. 2017, 16, 2623–2631. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.P.; Liu, A.; Weng, C.H.; Li, H.; Sun, S.; Song, A.; Zhu, H. Cloning and characterization of a novel phenylalanine ammonia-lyase gene from Inonotus baumii. Enzym. Microb. Technol. 2018, 112, 52–58. [Google Scholar] [CrossRef]
- Jacobs, A.T.; Marnett, L.J. Systems analysis of protein modification and cellular responses induced by electrophile stress. Accounts Chem. Res. 2010, 43, 673–683. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Andrade, M.O.; Gomes, A.P.; Damatta, F.M.; Baracat-Pereira, M.C.; Fontes, E.P. Arabidopsis and tobacco plants ectopically expressing the soybean antiquitin-like ALDH7 gene display enhanced tolerance to drought, salinity, and oxidative stress. J. Exp. Bot. 2006, 57, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Y.; Tan, Q.Q.; Jiang, S.X. First report of pine wilt disease caused by Bursaphelenchus xylophilus on Pinus thunbergii in the inland city of zibo, Shandong China. Plant Dis. 2013, 97, 1126. [Google Scholar] [CrossRef]
- Fang, S.M.; Wu, Z.R.; Qian, C.; Chen, C.F.; Li, J.; Sun, H.Q.; Shi, J. Effects of pine wood nematode disease on understory vegetation diversity and biomass. Fujian For. Sci. Technol. 2022, 49, 19–25+31. [Google Scholar]
- Wang, M.; Du, G.C.; Fang, J.N.; Wang, L.S.; Guo, Q.Q.; Zhang, T.T.; Li, R.G. UGT440A1 is associated with motility, reproduction, and pathogenicity of the plant-parasitic nematode Bursaphelenchus xylophilus. Front. Plant Sci. 2022, 13, 862594. [Google Scholar] [CrossRef] [PubMed]
- Proença, D.N.; Grass, G.; Morais, P.V. Understanding pine wilt disease: Roles of the pine endophytic bacteria and of the bacteria carried by the disease-causing pinewood nematode. MicrobiologyOpen 2017, 6, e00415. [Google Scholar] [CrossRef] [Green Version]
- Mannaa, M.; Han, G.; Jeon, H.W.; Kim, J.; Kim, N.; Park, A.R.; Kim, J.C.; Seo, Y.S. Influence of resistance-inducing chemical elicitors against pine wilt disease on the rhizosphere microbiome. Microorganisms 2020, 8, 884. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Kim, J.J.; Woo, K.S.; Jang, K.H.; Kim, Y.H.; Shim, D. De novo assembly and transcriptome analysis of the Pinus densiflora response to pine wilt disease in nature. Plant Biotechnol. Rep. 2018, 12, 229–236. [Google Scholar] [CrossRef]
- Santos, C.S.; Pinheiro, M.; Silva, A.I.; Egas, C.; Vasconcelos, M.W. Searching for resistance genes to Bursaphelenchus xylophilus using high throughput screening. BMC Genom. 2012, 13, 599. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.F.; Huang, A.Z.; Li, H.M.; Feng, L.Z.; Zhang, F.P.; Guo, W.S. Identification and comparative analysis of microRNAs in Pinus massoniana infected by Bursaphelenchus xylophilus. Plant Growth Regul. 2017, 83, 223–232. [Google Scholar] [CrossRef]
- McHale, L.; Tan, X.P.; Koehl, P.; Michelmore, R.W. Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 2006, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.E.; Coleman, M.J.; Szabo, V.; Frost, L.N.; Schmidt, R.; van der Biezen, E.A.; Moores, T.; Dean, C.; Daniels, M.J.; Jones, J.D. The Arabidopsis downy mildew resistance gene RPP5 shares similarity to the toll and interleukin-1 receptors with N and L6. Plant Cell 1997, 9, 879–894. [Google Scholar] [CrossRef] [Green Version]
- Radwan, O.; Mouzeyar, S.; Nicolas, P.; Bouzidi, M.F. Induction of a sunflower CC-NBS-LRR resistance gene analogue during incompatible interaction with Plasmopara halstedii. J. Exp. Bot. 2005, 56, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramkumar, G.; Madhav, M.; Biswal, A.; Devi, S.R.; Sakthivel, K.; Mohan, M.; Umakanth, B.; Mangrauthia, S.K.; Sundaram, R.; Viraktamath, B.C. Genome-wide identification and characterization of transcription factor binding motifs of NBS-LRR genes in Rice and Arabidopsis. J. Genomes Exomes 2014, 3, 7. [Google Scholar]
- Yang, S.H.; Zhang, X.H.; Yue, J.X.; Tian, D.; Chen, J.Q. Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol. Genet. Genom. 2008, 280, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, J.; Zhang, W.; Zhang, Y.; Tang, P.; Chen, J.Q.; Tian, D.; Yang, S. Unique evolutionary pattern of numbers of gramineous NBS-LRR genes. Mol. Genet. Genom. 2010, 283, 427–438. [Google Scholar] [CrossRef]
- Tong, C.; Zhang, Y.; Shi, F. Genome-wide identification and analysis of the NLR gene family in Medicago ruthenica. Front. Genet. 2023, 13, 1088763. [Google Scholar] [CrossRef]
- Lozano, R.; Hamblin, M.T.; Prochnik, S.; Jannink, J.L. Identification and distribution of the NBS-LRR gene family in the Cassava genome. BMC Genom. 2015, 16, 360. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.F.; Bi, C.Y.; Shi, Y.Y.; Hu, Y.Z.; Zhou, L.X.; Liang, C.X.; Huang, B.F.; Xu, M.; Lin, S.Q.; Chen, X.Y. Mining and analysis of NBS-LRR gene family in sweet potato genome. Acta. Crops Sin. 2020, 46, 1195–1207. [Google Scholar]
- Song, X.; Bai, S.H.; Dai, H.Y. Apple: Identification and expression analysis of NBS-LRR 1 gene. Acta. Horticultural. Sinica 2013, 411, 1233–1243. [Google Scholar]
- Zheng, Y.J.; Wu, Y.F.; Li, J.; Zhang, T.; Wang, X.D. Family analysis of NBS-LRR disease resistance gene family and CcRNL gene cloning in camphor tree. Biotechnol. Bull. 2018, 34, 142–149. [Google Scholar]
- Liu, Y.F.; Liu, L.J.; Yang, S.; Zeng, Q.; Liu, G.C.; Liu, Y.G. Cloning and expression analysis of NBS-LRR gene from spruce. Acta Bot. Boreal. Occident. Sin. 2020, 40, 1114–1122. [Google Scholar]
- Tylicki, A.; Siemieniuk, M. Thiamine and its derivatives in the regulation of cell metabolism. Postepy. Hig. Med. Dosw. 2011, 65, 447–469. [Google Scholar] [CrossRef] [PubMed]
- Coquille, S.; Roux, C.; Mehta, A.; Begley, T.P.; Fitzpatrick, T.B.; Thore, S. High-resolution crystal structure of the eukaryotic HMP-P synthase (THIC) from Arabidopsis thaliana. J. Struct. Biol. 2013, 184, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, W.Y.; Liao, J.C.; Wang, H.T.; Hung, T.H.; Tseng, C.C.; Chung, T.Y.; Hsieh, M.H. The Arabidopsis thiamin-deficient mutant pale green1 lacks thiamin monophosphate phosphatase of the vitamin B1 biosynthesis pathway. Plant J. 2017, 91, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Ajjawi, I.; Rodriguez Milla, M.A.; Cushman, J.; Shintani, D.K. Thiamin pyrophosphokinase is required for thiamin cofactor activation in Arabidopsis. Plant Mol. Biol. 2007, 65, 151–162. [Google Scholar] [CrossRef]
- Fitzpatrick, T.B.; Chapman, L.M. The importance of thiamine (vitamin B1) in plant health: From crop yield to biofortification. J. Biol. Chem. 2020, 295, 12002–12013. [Google Scholar] [CrossRef] [PubMed]
- Noordally, Z.; Trichtinger, C.; Dalvit, I.; Hofmann, M.; Roux, C.; Zamboni, N.; Pourcel, L.; Gas-Pascual, E.; Gisler, A.; Fitzpatrick, T. The coenzyme thiamine diphosphate displays a nuclear rhythm independent of the circadian clock. Commun. Biol. 2020, 3, 209. [Google Scholar] [CrossRef]
- Bocobza, S.; Adato, A.; Mandel, T.; Shapira, M.; Nudler, E.; Aharoni, A. Riboswitch-dependent gene regulation and its evolution in the plant kingdom. Genes Dev. 2007, 21, 2874–2879. [Google Scholar] [CrossRef] [Green Version]
- Hamada, A.M.; Jonsson, L.M.V. Thiamine treatments alleviate aphid infestations in barley and pea. Phytochemistry 2013, 94, 135–141. [Google Scholar] [CrossRef]
- Hamada, A.M.; Fatehi, J.; Jonsson, L.M.V. Seed treatments with thiamine reduce the performance of generalist and specialist aphids on crop plants. Bull. Entomol. Res. 2018, 108, 84–92. [Google Scholar] [CrossRef]
- Pushpalatha, H.G.; Sudisha, J.; Geetha, N.P.; Amruthesh, K.N.; Shekar Shetty, H. Thiamine seed treatment enhances LOX expression, promotes growth and induces downy mildew disease resistance in pearl millet. Biol. Plant 2011, 55, 522–527. [Google Scholar] [CrossRef]
- Bahuguna, R.N.; Joshi, R.; Shukla, A.; Pandey, M.; Kumar, J. Thiamine primed defense provides reliable alternative to systemic fungicide carbendazim against sheath blight disease in rice (Oryza sativa L.). Plant Physiol. Bioch. 2012, 57, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.P.; Kim, S.; Lee, Y.H. Vitamin B1 functions as an activator of plant disease resistance. Plant Physiol. 2005, 138, 1505–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.L.; Wang, Y.H.; Wang, T.Y.; Zhu, Y.; Wu, P.; Li, L.J. Comparative metabolomic profiling of secondary metabolites in different tissues of Euryale ferox and functional characterization of phenylalanine ammonia-lyase. Ind. Crops Prod. 2023, 195, 116450. [Google Scholar] [CrossRef]
- Chen, Y.K. Study on genetic transformation of rice with Phenylalanine Ammonia-lyase. Fujian Agric. For. Univ. 2011, 7, 1–66. [Google Scholar]
- Xie, B.X.; Zhang, M.H. The recent advances on the function and application of flavonoids. Acta Zoonutrimenta Sin. 2003, 15, 11–15. [Google Scholar]
- Shiragaki, K.; Nakamura, R.; Nomura, S.; He, H.; Yamada, T.; Marubashi, W.; Oda, M.; Tezuka, T. Phenylalanine ammonia-lyase and phenolic compounds are related to hybrid lethality in the cross Nicotiana suaveolens × N. tabacum. Plant Biotechnol. 2020, 37, 327–333. [Google Scholar] [CrossRef]
- Ren, Q.; Hu, Y.J.; Li, Z.Y.; Jin, Y.J. Lignin content and peroxidase activity of Pinus massoniana. Acta Ecol. Sin. 2007, 11, 4895–4899. [Google Scholar]
- Yang, Y.W.; Li, S.; Huang, J.Y.; Zhang, B.L. Identification and analysis of the gene family of phenylalanine ammonialyase in upland cotton. Mol. Plant Breed. 2017, 15, 1184–1191. [Google Scholar]
- Qiao, F.; Geng, G.G.; Zhang, L.; Jin, L.; Chen, Z. Molecular cloning and expression patterns of LcPAL from Lycium chinense. J. China Agric. Univ. 2017, 22, 64–73. [Google Scholar]
- Sun, Y.J.; Chen, X.; Cui, H.N.; Wu, C.; Liu, S.; Luan, F.S.; Wang, X.Z. Bioinformatics analysis of PAL gene family in cucurbitaceae and the PAL4 gene cloning in cucumis melo L. Mol. Plant Breed 2018, 16, 4910–4920. [Google Scholar]
- Chen, Y.H.; Ye, J.R.; Wei, C.J. Effect of pine wood nematode (PWN) infection on phenylpropanes metabolism in masson pine seedlings. Sci. Silv. Sin. 2006, 2, 73–77. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Data (bp) | Clean Data (bp) | AF_Q20 (%) | AF_Q30 (%) | AF_GC (%) |
|---|---|---|---|---|---|
| CK-1 | 7,154,388,900 | 7,097,563,080 | 98.04 | 94.32 | 45.13 |
| CK-2 | 7,086,201,600 | 7,030,845,840 | 98.12 | 94.52 | 45.24 |
| CK-3 | 7,342,288,200 | 7,287,440,931 | 98.00 | 94.23 | 45.10 |
| BL-1 | 7,096,215,600 | 7,039,809,015 | 97.84 | 93.80 | 45.35 |
| BL-2 | 6,642,519,000 | 6,587,501,867 | 98.08 | 94.36 | 45.36 |
| BL-3 | 7,055,390,400 | 6,993,540,884 | 98.02 | 94.28 | 45.21 |
| ID | Primer Name | Sequence (5′to 3′) |
|---|---|---|
| Unigene0001984 | Unigene0001984-F | CAATCAATGGAGCAGTAGA |
| Unigene0001984-R | GCAATAGGTGGTGTAATGA | |
| Unigene0083685 | Unigene0083685-F | CATCTTCTGCTGTTCAATC |
| Unigene0083685-R | GCTCTTCATCAAAGTTACC | |
| Unigene0033503 | Unigene0033503-F | GGATTCTACACTTACAATGC |
| Unigene0033503-R | CATAACATTAGCGAAGAAGG | |
| Unigene0000278 | Unigene0000278-F | CGCTGGTTGAATATACTCT |
| Unigene0000278-R | GGATATAGGTAGGCATCTG | |
| Unigene0048705 | Unigene0048705-F | CTTGTAACAGCAGAGGAG |
| Unigene0048705-R | CAATACCAGAGAGGGAAAT | |
| Unigene0083090 | Unigene0083090-F | CCTCTTGAATTGCCTCAT |
| Unigene0083090-R | GACCAACAACCATTATGC | |
| Unigene0003367 | Unigene0003367-F | CTGAAAGTGACTGATGGA |
| Unigene0003367-R | TTGGAATAGTTCGTAGGAG | |
| Unigene0035857 | Unigene0035857-F | GCTTCTGGACTCTTTCAA |
| Unigene0035857-R | CTGTCTGTTCTCCTCCTA | |
| Unigene0005880 | Unigene0005880-F | ACCTGAGAAGAAGAAGATTC |
| Unigene0005880-R | GCAAAGAGCCTATGGATT | |
| Unigene0020248 | Unigene0020248-F | TGCTCTGGCTTCCATCGTTT |
| Unigene0020248-R | CTGGTTTCCCGAGCATCACT | |
| Unigene0074724 | Unigene0074724-F | ATGCGAGGGATTGCTCCTTG |
| Unigene0074724-R | CTGTGGAGCCCCATGAACTG | |
| Unigene0012357 | Unigene0012357-F | CGTATGAGCCCCAGTATGCC |
| Unigene0012357-R | GGTCCCATCTTCTCACAGCC | |
| U2af | U2af-F | TCGGGAGGTTGGGTCTACAT |
| U2af-R | ACCAGTCCTTCAGTCCCCTT |
| ID | log2(fc) | Description |
|---|---|---|
| Unigene0059253 | 1.6229304 | TIR/NBS/LRR disease resistance protein |
| Unigene0007068 | 5.6885001 | TIR-NBS-LRR protein |
| Unigene0035790 | 1.6295281 | TIR-NBS-LRR protein |
| Unigene0043826 | 1.9064777 | LRR receptor-like serine/threonine-protein kinase EFR |
| Unigene0069489 | 4.5993619 | LRR receptor-like serine/threonine-protein kinase At3g47570 |
| Unigene0031688 | 7.5062084 | LRR receptor-like serine/threonine-protein kinase At1g56140 isoform X1 |
| Unigene0003092 | 10.81645 | LRR receptor-like serine/threonine-protein kinase At3g47570 |
| Unigene0085494 | 10.70275 | LRR receptor-like serine/threonine-protein kinase At3g47570 |
| Unigene0002084 | −2.057195 | LRR receptor-like serine/threonine-protein kinase At3g47570 isoform X6 |
| TF Families | Description |
|---|---|
| ERF | unigene0084830, unigene0028109, unigene0072376 |
| MYB | unigene0100697, unigene0040542, unigene0073218 |
| WRKY | unigene0046254 |
| ID | log2(fc) | Description |
|---|---|---|
| Unigene0010372 | 11.859793 | Putative thiamine pyrophosphokinase |
| Unigene0020296 | 12.303019 | Phosphomethylpyrimidine synthase |
| Unigene0027776 | 10.550746 | Phosphomethylpyrimidine synthase |
| Unigene0060145 | 12.404964 | Thiamine phosphate phosphatase/amino-HMP aminohydrolase |
| Unigene0089674 | 6.915879 | Thiamine phosphate phosphatase/amino-HMP aminohydrolase |
| Unigene0020248 | −1.546972 | Phenylalanine ammonia-lyase |
| Unigene0079836 | 10.090112 | Phenylalanine ammonia-lyase |
| Unigene0010629 | −1.785339 | Aldehyde dehydrogenase family 2 member B7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Du, G.; Guo, Q.; Dong, G.; Wang, M.; Zhang, T.; Li, R. Transcriptome Sequencing and Analysis of Genes Related to Disease Resistance in Pinus thunbergii. Forests 2023, 14, 650. https://doi.org/10.3390/f14030650
Zhang Y, Du G, Guo Q, Dong G, Wang M, Zhang T, Li R. Transcriptome Sequencing and Analysis of Genes Related to Disease Resistance in Pinus thunbergii. Forests. 2023; 14(3):650. https://doi.org/10.3390/f14030650
Chicago/Turabian StyleZhang, Yu, Guicai Du, Qunqun Guo, Guosong Dong, Min Wang, Tingting Zhang, and Ronggui Li. 2023. "Transcriptome Sequencing and Analysis of Genes Related to Disease Resistance in Pinus thunbergii" Forests 14, no. 3: 650. https://doi.org/10.3390/f14030650
APA StyleZhang, Y., Du, G., Guo, Q., Dong, G., Wang, M., Zhang, T., & Li, R. (2023). Transcriptome Sequencing and Analysis of Genes Related to Disease Resistance in Pinus thunbergii. Forests, 14(3), 650. https://doi.org/10.3390/f14030650