
Physiological Differences and Transcriptome Analysis Reveal That High Enzyme Activity Significantly Enhances Drought Tolerance in Chinese Fir (Cunninghamia lanceolata)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Screening of Study Material with Different Drought-Tolerance Capacities
2.1.1. Screening Experimental Design
2.1.2. Determination of Leaf Relative Water Content, Plasma Membrane Permeability, and Lipid Peroxidation
2.1.3. Data Analysis
2.2. Physiological Response of Different DT Clones to Drought Stress
2.2.1. Drought Stress Experimental Design
2.2.2. Determinations of Antioxidant Enzyme Activity
2.2.3. Data Statistics
2.3. Transcriptome Analysis
2.3.1. RNA Extraction and Inspection
2.3.2. cDNA Library Preparation and Transcriptome Sequencing
2.3.3. Analysis of Illumina Transcriptome Sequencing Results
2.3.4. Bioinformatics for Functional Annotation of Differentially Expressed Genes
2.3.5. GO Functional and KEGG Pathway Enrichment Analysis for DEGs
3. Results
3.1. Screening of Different Drought-Tolerant Chinese Fir Clones
3.2. Effects of Drought Stress on Antioxidant Enzyme Activity among High and Low DT Clones
3.3. RNA-Sequencing and Read Assembly
3.4. Gene Annotation and Functional Classification
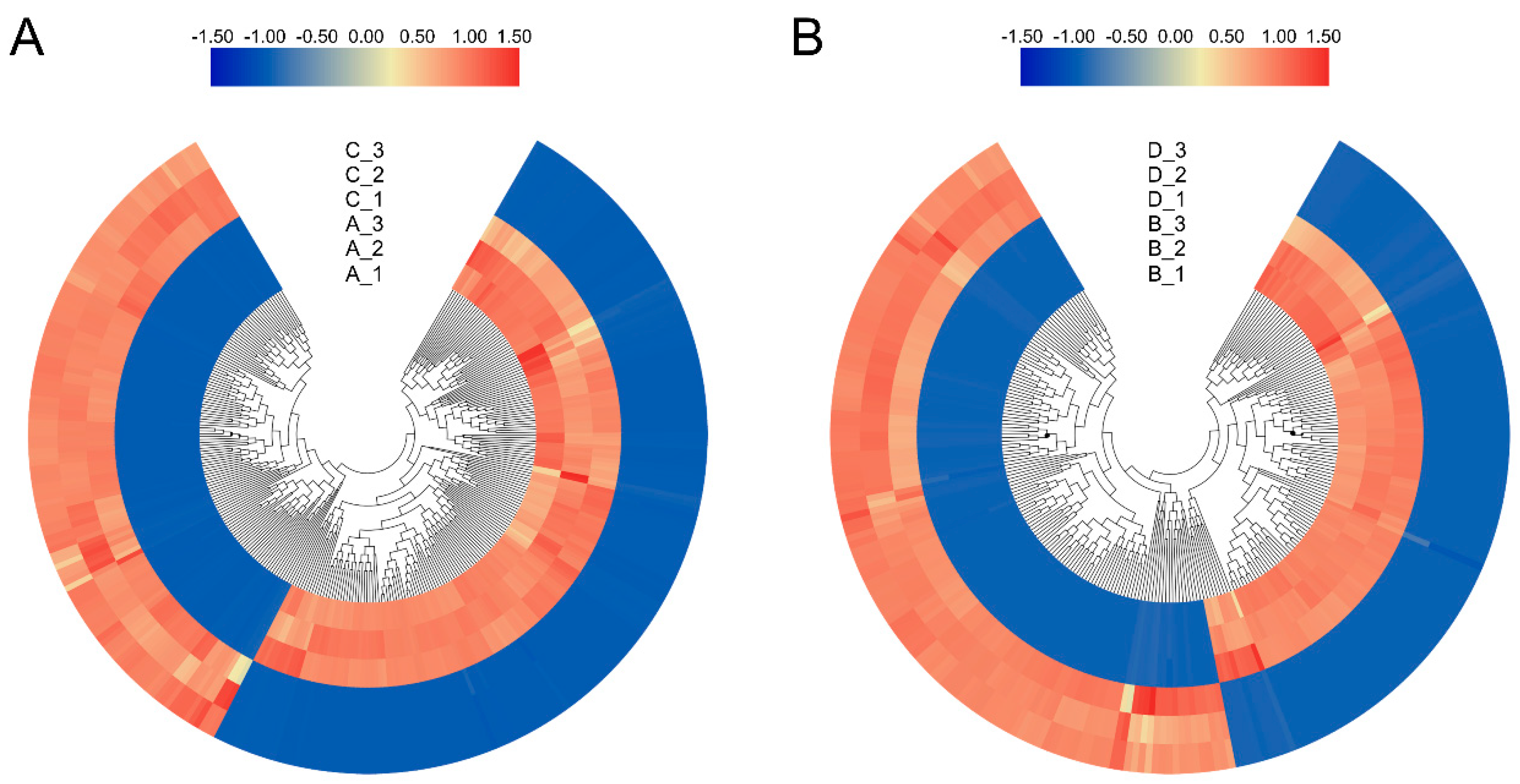
3.5. Differently Expressed Genes (DEGs) under Drought Stress
3.6. Gene Enrichment Analysis for DEGs among Four Libraries
3.7. Key DEGs in Response to Drought Stress in Chinese Fir
3.8. ClCHIs and ClPERs Highly Expressed in No. 228 Clone
4. Discussion
4.1. Differences Responses of Antioxidant Enzyme among High and Low DT Chinese Fir Clones
4.2. Transcriptome Sequencing and Annotation
4.3. Key Enriched DEG Categories in GO and KEGG Pathways
4.4. Transcription Factors Participate in Plant Responses to Drought Stress
4.5. Expression of Auxin and ABA Response Genes under Drought Stress
4.6. Expression of Gene Encoding Protein Kinase under Drought Stress
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ploughe, L.W.; Jacobs, E.M.; Frank, G.S.; Greenler, S.M.; Smith, M.D.; Dukes, J.S. Community Response to Extreme Drought (CRED): A framework for drought–induced shifts in plant–plant interactions. New Phytol. 2019, 222, 52–69. [Google Scholar] [CrossRef]
- Choat, B.; Brodribb, T.J.; Brodersen, C.R.; Duursma, R.A.; López, R.; Medlyn, B.E. Triggers of tree mortality under drought. Nature 2018, 558, 531–539. [Google Scholar] [CrossRef]
- Noctor, G.; Mhamdi, A.; Foyer, C.H. The roles of reactive oxygen metabolism in drought: Not so cut and dried. Plant Physiol. 2014, 164, 1636–1648. [Google Scholar] [CrossRef]
- Fang, Y.; Xiong, L. General mechanisms of drought response and their application in drought resistance improvement in plants. Cell Mol. Life Sci. 2015, 72, 673–689. [Google Scholar] [CrossRef]
- Cabello, J.V.; Lodeyro, A.F.; Zurbriggen, M.D. Novel perspectives for the engineering of abiotic stress tolerance in plants. Curr. Opin. Biotechnol. 2014, 26, 62–70. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Nahar, K.; Hossain, M.S.; Mahmud, J.A.; Rahman, A.; Inafuku, M.; Oku, H.; Fujita, M. Coordinated actions of glyoxalase and antioxidant defense systems in conferring abiotic stress tolerance in plants. Int. J. Mol Sci. 2017, 18, 200. [Google Scholar] [CrossRef]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Cruz, D.C.M. Drought stress and reactive oxygen species: Production, scavenging and signaling. Plant Signal Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef]
- Reguera, M.; Peleg, Z.; Blumwald, E. Targeting metabolic pathways for genetic engineering abiotic stress–tolerance in crops. Biochim. Biophys Acta 2012, 1819, 186–194. [Google Scholar] [CrossRef]
- Sarwat, M.; Naqvi, A.R. Heterologous expression of rice calnexin (OsCNX) confers drought tolerance in Nicotiana tabacum. Mol. Biol. Rep. 2013, 40, 5451–5464. [Google Scholar] [CrossRef]
- Li, Z.Q.; Li, J.T.; Bing, J.; Zhang, G.F. Analysis of the role of Arabidopsis APX family genes in plant growth and development and response to abiotic stress. Genet 2019, 41, 534–549. (In Chinese) [Google Scholar]
- Liu, J.X.; Feng, K.; Duan, A.Q.; Li, H.; Yang, Q.Q.; Xu, Z.S.; Xiong, A.S. Isolation, purification and characterization of an ascorbate peroxidase from celery and overexpression of the AgAPX1 gene enhanced ascorbate content and drought tolerance in Arabidopsis. BMC Plant Biol. 2019, 19, 488. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, M.; Teng, Y.; Jia, S.; Yu, D.; Wei, T.; Chen, C.; Song, W. Overexpression of the glutathione peroxidase 5 (RCGPX5) gene from rhodiola crenulata increases drought tolerance in Salvia miltiorrhiza. Front. Plant Sci. 2018, 9, 1950. [Google Scholar] [CrossRef]
- Llorente, F.; López–Cobollo, R.M.; Catalá, R.; Martínez–Zapater, J.M.; Salinas, J. A novel cold–inducible gene from Arabidopsis, RCI3, encodes a peroxidase that constitutes a component for stress tolerance. Plant J. 2002, 32, 13–24. [Google Scholar] [CrossRef]
- Feng, K.; Yu, J.; Cheng, Y.; Ruan, M.; Wang, R.; Ye, Q.; Zhou, G.; Li, Z.; Yao, Z.; Yang, Y.; et al. The SOD Gene Family in Tomato: Identification, Phylogenetic Relationships, and Expression Patterns. Front. Plant Sci. 2016, 7, 1279. [Google Scholar] [CrossRef]
- Molina–Rueda, J.J.; Tsai, C.J.; Kirby, E.G. The Populus superoxide dismutase gene family and its responses to drought stress in transgenic poplar overexpressing a pine cytosolic glutamine synthetase (GS1a). PLoS ONE. 2013, 8, e56421. [Google Scholar] [CrossRef]
- Wang, F.Z.; Wang, Q.B.; Kwon, S.Y.; Kwak, S.S.; Su, W.A. Enhanced drought tolerance of transgenic rice plants expressing a pea manganese superoxide dismutase. J. Plant Physiol. 2005, 162, 465–472. [Google Scholar] [CrossRef]
- Phung, T.H.; Jung, H.I.; Park, J.H.; Kim, J.G.; Back, K.; Jung, S. Porphyrin biosynthesis control under water stress: Sustained porphyrin status correlates with drought tolerance in transgenic rice. Plant Physiol. 2011, 157, 1746–1764. [Google Scholar] [CrossRef]
- Zhou, L.L.; Li, S.B.; Liu, B.; Wu, S.P.; Heal, K.V.; Ma, X.Q. Tissue–specific carbon concentration, carbon stock, and distribution in Cunninghamia lanceolata (Lamb.) Hook plantations at various developmental stages in subtropical China. Ann. For. Sci. 2019, 76, 70. [Google Scholar] [CrossRef]
- Farooq, T.H.; Yan, W.; Rashid, M.H.U. Chinese fir (Cunninghamia lanceolata) a green gold of China with continues decline in its productivity over the successive rotations: A review. Appl. Ecol. Environ. Res. 2019, 17, 11055–11067. [Google Scholar] [CrossRef]
- Song, X.; Yu, G.R.; Liu, Y.F.; Sun, X.M.; Lin, Y.M.; Wen, X.F. Seasonal changes in water use efficiency of subtropical plantations and their impact on environmental factors. Sci in China. Ser. D-Ear. Sci. 2006, 49, 111–118. (In Chinese) [Google Scholar]
- Lin, Y.L. Effects of the 2003 drought on the growth of 1- to 3-year-old Chinese fir (Cunninghamia Lanceolata) plantation. J. Fujian For. Sci. Tec. 2004, 31, 31–34. (In Chinese) [Google Scholar]
- Dong, T.F.; Duan, B.L.; Zhang, S.; Korpelainen, H.; Niinemets, Ü.; Li, C.Y. Growth, biomass allocation and photosynthetic responses are related to intensity of root severance and soil moisture conditions in the plantation tree Cunninghamia lanceolata. Tree Physiol. 2016, 36, 807–817. [Google Scholar] [CrossRef]
- Li, S.B.; Zhou, L.L.; Wu, S.P.; Sun, M.; Ding, G.C.; Lin, S.Z. Effects of different nitrogen forms on nutrient absorption and distribution of Chinese fir seedlings under drought stress. J. Plant Nutr. Fert. 2020, 26, 152–162. (In Chinese) [Google Scholar]
- Li, S.B.; Zhou, L.L.; Addo–Danso, S.D.; Ding, G.C.; Sun, M.; Wu, S.P.; Lin, S.Z. Nitrogen supply enhances the physiological resistance of Chinese fir plantlets under polyethylene glycol (PEG)–induced drought stress. Sci. Rep. 2020, 10, 7509. [Google Scholar] [CrossRef]
- Li, J.H.; Luo, D.W.; Ma, G.F.; Jia, L.C.; Xu, J.L.; Huang, H.H.; Tong, Z.K.; Lu, Y.Q. Response of Chinese fir seedlings to low phosphorus stress and analysis of gene expression differences. J. For. Res. 2018, 30, 183–192. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Liu, W.; Luo, Z.; Wang, P.; Zhang, Y.; Zheng, R.; Shi, J. Transcriptome characteristics and six alternative expressed genes positively correlated with the phase transition of annual cambial activities In Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook). PLoS ONE 2013, 8, e71562. [Google Scholar] [CrossRef]
- Hu, R.; Wu, B.; Zheng, H.; Hu, D.; Wang, X.; Duan, H.; Sun, Y.; Wang, J.; Zhang, Y.; Li, Y. Global reprogramming of transcriptionIn Chinese fir (Cunninghamia Lanceolata) during progressive drought stress and after rewatering. Int. J. Mol. Sci. 2015, 16, 15194–15219. [Google Scholar] [CrossRef]
- Liu, H.; Sultan, M.A.; Liu, X.L.; Zhang, J.; Yu, F.; Zhao, H.X. Physiological and comparative proteomic analysis reveals different drought responses in roots and leaves of drought–tolerant wild wheat (Triticum boeoticum). PLoS ONE 2015, 10, e121852. [Google Scholar] [CrossRef]
- Li, S.B.; Ding, G.C.; Cao, G.Q.; Lin, S.Z.; Guo, W.Z. Comprehensive evaluation of drought resistance of different Chinese fir clones under simulated stress. J. Fujian Agri. Univ. (Nat. Sci. Ed.) 2012, 41, 491–496. (In Chinese) [Google Scholar]
- Yang, S.; Xu, K.; Chen, S.; Li, T.; Xia, H.; Chen, L.; Liu, H.; Luo, L. A stress–responsive bZIP transcription factor OsbZIP62 improves drought and oxidative tolerance in rice. BMC Plant Biol. 2019, 19, 260. [Google Scholar] [CrossRef]
- Li, J.J.; Yu, J.H.; Chang, Y.J.; Xu, X.Y.; Nie, S.M. Effects of high temperature stress on plasma membrane permeability and protective enzyme activities of cucumber seedling leaves. J. Changjiang Veg. 2007, 24, 59–61. (In Chinese) [Google Scholar]
- Qin, P.; Liu, Y.J.; Liu, F.H. Effects of drought stress on malondialdehyde content and cell membrane permeability of tobacco leaves. Subtrop. Plant Sci. 2004, 12, 8–10. (In Chinese) [Google Scholar]
- Long, H.T.; Li, L.M.; Xie, Z.H.; Liu, S.; Li, X.Y.; Deng, B.; Liu, H.Y.; Li, L. Evaluation of the relationship between drought resistance of peanut varieties and AhNCED1 gene expression by comprehensive membership function method. J. Integr. Plant Biol. 2015, 50, 706–712. (In Chinese) [Google Scholar]
- Shan, Z.; Luo, X.; Wei, M.; Huang, T.; Khan, A.; Zhu, Y. Physiological and proteomic analysis on long-term drought resistance of cassava (Manihot esculenta Crantz). Sci. Rep. 2018, 8, 17982. [Google Scholar] [CrossRef]
- Yang, J.P.; Jin, W.L.; Xu, H.M.; Wang, W.P.; Chen, Y.Y. Influence of SA on POD in Leaves of Red Bean under Water Stress. J. Beijing Agri. Coll. 2002, 9, 11–14. [Google Scholar]
- Si, H.Q.; Ma, C.X.; He, K.Q.; Zhou, N.; Hu, N. Comparative Analysis on Detection Methods for Activity of Polyphenol Oxidase in Wheat. Cereal Feed. Indus. 2007, 1, 8–10. (In Chinese) [Google Scholar]
- Li, Y.Q.; Song, Z.W.; Wang, L.; Jin, Z.Y.; Yuan, H.J. Effect of spider mite damage on the activities of superoxide dismutase and catalase in cowpea leaves. J. Southwest China Norm. Univ. (Nat. Sci. Ed.) 2001, 3, 62–65. (In Chinese) [Google Scholar]
- Mu, Z.Z.; Keremu, Y.L.; Wang, Y.J. Effects of high temperature and drought on leaf physiological indexes of Korla fragrant pear. Jiangsu Agr. Sci. 2016, 44, 209–212. (In Chinese) [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Moran, Y.; Levin, J.Z.; Thompson, D.A.; Ido, A.; Xian, A.; Lin, F.; Raktima, R.; Zeng, Q.D.; et al. Full–length transcriptome assembly from RNA–Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [PubMed]
- Götz, S.; García–Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High–throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Yoav, B.; Daniel, Y. The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann. Stat. 2001, 4, 1165–1188. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA–seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Shabala, L.; Brodribb, T.J.; Zhou, M.; Shabala, S. Assessing the suitability of various screening methods as a proxy for drought tolerance in barley. Funct. Plant Biol. 2017, 44, 253–266. [Google Scholar] [CrossRef]
- Gholami, M.; Rahemi, M.; Rastegar, S. Use of rapid screening methods for detecting drought tolerant cultivars of fig (Ficus carica L.). Sci. Hortic. 2012, 143, 7–14. [Google Scholar] [CrossRef]
- Ansari, W.A.; Atri, N.; Ahmad, J.; Qureshi, M.I.; Singh, B.; Kumar, R.; Rai, V.; Pandey, S. Drought mediated physiological and molecular changes in muskmelon (Cucumis melo L.). PLoS ONE 2019, 14, e222647. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, W.H.; Zhou, J.Y.; Ma, C.; Han, W.J. Effects of drought stress on the growth and physiological and biochemical indexes of fine roots of Quercus variabilis seedlings. J. Ecol. 2014, 34, 4223–4233. (In Chinese) [Google Scholar]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High–throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9, 312. [Google Scholar] [CrossRef]
- Qiu, T.; Chen, Y.; Li, M.; Kong, Y.; Zhu, Y.; Han, N.; Bian, H.; Zhu, M.; Wang, J. The tissue–specific and developmentally regulated expression patterns of the SAUR41 subfamily of small AUXIN UP RNA genes. Plant Signal Behav. 2013, 8, e25283. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.H.; Dong, C.M.; Zheng, X.K.; Feng, W.S.; Liu, M.Q.; Zhao, L. Transcriptome analysis reveals genes involved in biosynthesis of secondary metabolism in Cornus officinalis. Chin. J. Chin. Mat Medic. 2017, 42, 213–219. (In Chinese) [Google Scholar]
- Wang, X.; Chen, D.; Wang, Y.; Xie, J. De novo transcriptome assembly and the putative biosynthetic pathway of steroidal sapogenins of Dioscorea composita. PLoS ONE 2015, 10, e124560. [Google Scholar] [CrossRef]
- Arya, S.K.; Dhar, Y.V.; Upadhyay, S.K.; Asif, M.H.; Verma, P.C. De novo characterization of Phenacoccus solenopsis transcriptome and analysis of gene expression profiling during development and hormone biosynthesis. Sci. Rep. 2018, 8, 7573. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Zhang, Y.; Song, J.; Zhao, L.; Wang, Z. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 2011, 98, 272–279. [Google Scholar]
- Iorizzo, M.; Senalik, D.A.; Grzebelus, D.; Bowman, M.; Cavagnaro, P.F.; Matvienko, M.; Ashrafi, H.; Van Deynze, A.; Simon, P.W. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genom. 2011, 12, 389. [Google Scholar] [CrossRef]
- Zeng, X.; Bai, L.; Wei, Z.; Yuan, H.; Wang, Y.; Xu, Q.; Tang, Y.; Nyima, T. Transcriptome analysis revealed the drought–responsive genes in Tibetan hulless barley. BMC Genom. 2016, 17, 386. [Google Scholar] [CrossRef]
- Dong, Y.; Fan, G.; Zhao, Z.; Deng, M. Transcriptome expression profiling in response to drought stress in Paulownia australis. Int. J. Mol. Sci. 2014, 15, 4583–4607. [Google Scholar] [CrossRef]
- Fox, H.; Doron–Faigenboim, A.; Kelly, G.; Bourstein, R.; Attia, Z.; Zhou, J.; Moshe, Y.; Moshelion, M.; David–Schwartz, R. Transcriptome analysis of Pinus halepensis under drought stress and during recovery. Tree Physiol. 2018, 38, 423–441. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, B.L.; Ma, C.; Zhang, G.; Chang, J.; Si, H.; Wang, D. Transcriptome characterization and sequencing–based identification of drought–responsive genes in potato. Mol. Biol. Rep. 2014, 41, 505–517. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, F.; Zhang, F.; Wang, W.; Zhou, Y.; Fu, B.; Li, Z. Comparative transcriptome sequencing of tolerant rice introgression line and its parents in response to drought stress. BMC Genom. 2014, 15, 1026. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [PubMed]
- Dash, M.; Yordanov, Y.S.; Georgieva, T.; Tschaplinski, T.J.; Yordanova, E.; Busov, V. Poplar PtabZIP1–like enhances lateral root formation and biomass growth under drought stress. Plant J. 2017, 89, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Li, S.; An, X.; Liu, X.; Qin, H.; Wang, D. Transgenic expression of MYB15 confers enhanced sensitivity to abscisic acid and improved drought tolerance in Arabidopsis thaliana. J. Genet. Genom. 2009, 36, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Seo, P.J.; Xiang, F.; Qiao, M.; Park, J.-Y.; Lee, Y.N.; Kim, S.-J.; Lee, Y.H.; Park, W.J.; Park, C.-M. The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis. Plant Physiol. 2009, 151, 275–289. [Google Scholar] [CrossRef]
- Urao, T.; Noji, M.; Yamaguchi–Shinozaki, K.; Shinozaki, K. A transcriptional activation domain of ATMYB2, a drought–inducible Arabidopsis MYB–related protein. Plant J. 1996, 10, 1145–1148. [Google Scholar] [CrossRef]
- Qiu, Y.; Yu, D. Over–expression of the stress–induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis. Environ. Exp. Bot. 2009, 65, 35–47. [Google Scholar] [CrossRef]
- Wu, X.; Shiroto, Y.; Kishitani, S.; Ito, Y.; Toriyama, K. Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep. 2009, 28, 21–30. [Google Scholar] [CrossRef]
- Zhou, M.; Ma, J.; Zhao, Y.; Wei, Y.; Tang, Y.; Wu, Y. Improvement of drought and salt tolerance in Arabidopsis and Lotus corniculatus by overexpression of a novel DREB transcription factor from Populus euphratica. Gene 2012, 506, 10–17. [Google Scholar] [CrossRef]
- Wei, T.; Deng, K.; Liu, D.; Gao, Y.; Liu, Y.; Yang, M.; Zhang, L.; Zheng, X.; Wang, C.; Song, W.; et al. Ectopic Expression of DREB Transcription Factor, AtDREB1A, Confers Tolerance to Drought in Transgenic Salvia miltiorrhiza. Plant Cell Physiol. 2016, 57, 1593–1609. [Google Scholar] [CrossRef]
- Kinoshita, N.; Wang, H.; Kasahara, H.; Liu, J.; Macpherson, C.; Machida, Y.; Kamiya, Y.; Hannah, M.A.; Chua, N.H. IAA–Ala Resistant 3, an evolutionarily conserved target of miR167, mediates Arabidopsis root architecture changes during high osmotic stress. Plant Cell. 2012, 24, 3590–3602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, J.; Zhang, W.; Yan, S.; Wang, R.; Zhao, J.; Li, Y.; Qi, Z.; Sun, Z.; Zhu, Z. The putative auxin efflux carrier OsPIN3t is involved in the drought stress response and drought tolerance. Plant J. 2012, 72, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhu, Y.; Gao, C.; She, W.; Lin, W.; Chen, Y.; Han, N.; Bian, H.; Zhu, M.; Wang, J. Tissue–specific expression of SMALL AUXIN UP RNA41 differentially regulates cell expansion and root meristem patterning in Arabidopsis. Plant Cell Physiol. 2013, 54, 609–621. [Google Scholar] [CrossRef]
- Qiu, Z.; Wan, L.; Chen, T.; Wan, Y.; He, X.; Lu, S.; Wang, Y.; Lin, J. The regulation of cambial activity In Chinese fir (Cunninghamia lanceolata) involves extensive transcriptome remodeling. New Phytol. 2013, 199, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Rivero, R.M.; Shulaev, V.; Blumwald, E.; Mittler, R. Abiotic and biotic stress combinations. New Phytol. 2014, 203, 32–43. [Google Scholar] [CrossRef]
- Du, H.; Huang, F.; Wu, N.; Li, X.; Hu, H.; Xiong, L. Integrative Regulation of Drought Escape through ABA–Dependent and –Independent Pathways in Rice. Mol. Plant. 2018, 11, 584–597. [Google Scholar] [CrossRef]
- Fujita, Y.; Fujita, M.; Satoh, R.; Maruyama, K.; Parvez, M.M.; Seki, M.; Hiratsu, K.; Ohme–Takagi, M.; Shinozaki, K.; Yamaguchi–Shinozaki, K. AREB1 is a transcription activator of novel ABRE–dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell. 2005, 17, 3470–3488. [Google Scholar] [CrossRef]
- Yoshida, T.; Mogami, J.; Yamaguchi–Shinozaki, K. ABA–dependent and ABA–independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef]
- Yu, L.X.; Djebrouni, M.; Chamberland, H.; Lafontaine, J.G.; Tabaeizadeh, Z. Chitinase: Differential induction of gene expression and enzyme activity by drought stress in the wild (Lycopersicon chilense Dun.) and cultivated (L. esculentum Mill.) tomatoes. J. Plant Physiol. 1998, 153, 745–753. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Values |
|---|---|
| Total number of raw reads | 880,448,444 |
| Total number of clean reads | 863,141,178 |
| Total nucleotide length | 129.47 Gb |
| Total number of unigenes | 380,835 |
| Mean length of unigenes | 1125 bp |
| Q20 percentage (%) | 96.90 |
| Q30 percentage (%) | 91.95 |
| GC percentage (%) | 44.49 |
| Sequence Database | Number of Annotated Unigenes | Percentage of Annotated Unigene (%) |
|---|---|---|
| NR | 188,114 | 49.39 |
| NT | 73,490 | 19.29 |
| KO | 70,927 | 18.62 |
| Swiss-Prot | 153,508 | 40.30 |
| PFAM | 163,178 | 42.84 |
| GO | 164,930 | 43.3 |
| KOG | 49,395 | 12.97 |
| Annotated in all Databases | 19,497 | 5.11 |
| Annotated in at least one Database | 227,019 | 59.61 |
| Total unigenes | 380,835 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Yan, X.; Huang, X.; Addo-Danso, S.D.; Lin, S.; Zhou, L. Physiological Differences and Transcriptome Analysis Reveal That High Enzyme Activity Significantly Enhances Drought Tolerance in Chinese Fir (Cunninghamia lanceolata). Forests 2023, 14, 967. https://doi.org/10.3390/f14050967
Li S, Yan X, Huang X, Addo-Danso SD, Lin S, Zhou L. Physiological Differences and Transcriptome Analysis Reveal That High Enzyme Activity Significantly Enhances Drought Tolerance in Chinese Fir (Cunninghamia lanceolata). Forests. 2023; 14(5):967. https://doi.org/10.3390/f14050967
Chicago/Turabian StyleLi, Shubin, Xinyang Yan, Xiaoyan Huang, Shalom Daniel Addo-Danso, Sizu Lin, and Lili Zhou. 2023. "Physiological Differences and Transcriptome Analysis Reveal That High Enzyme Activity Significantly Enhances Drought Tolerance in Chinese Fir (Cunninghamia lanceolata)" Forests 14, no. 5: 967. https://doi.org/10.3390/f14050967
APA StyleLi, S., Yan, X., Huang, X., Addo-Danso, S. D., Lin, S., & Zhou, L. (2023). Physiological Differences and Transcriptome Analysis Reveal That High Enzyme Activity Significantly Enhances Drought Tolerance in Chinese Fir (Cunninghamia lanceolata). Forests, 14(5), 967. https://doi.org/10.3390/f14050967