
Comparative Analyses of Acacia Plastomes to Detect Mutational Hotspots and Barcode Sites for the Identification of Important Timber Species
 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Samples and DNA Extraction
2.2. Genome Sequencing and Assembly
2.3. Genome Annotation
2.4. Repetitive DNA Analyses
2.5. Nucleotide Diversity
2.6. Phylogenetic Analyses and Nucleotide Substitutions
3. Results and Discussion
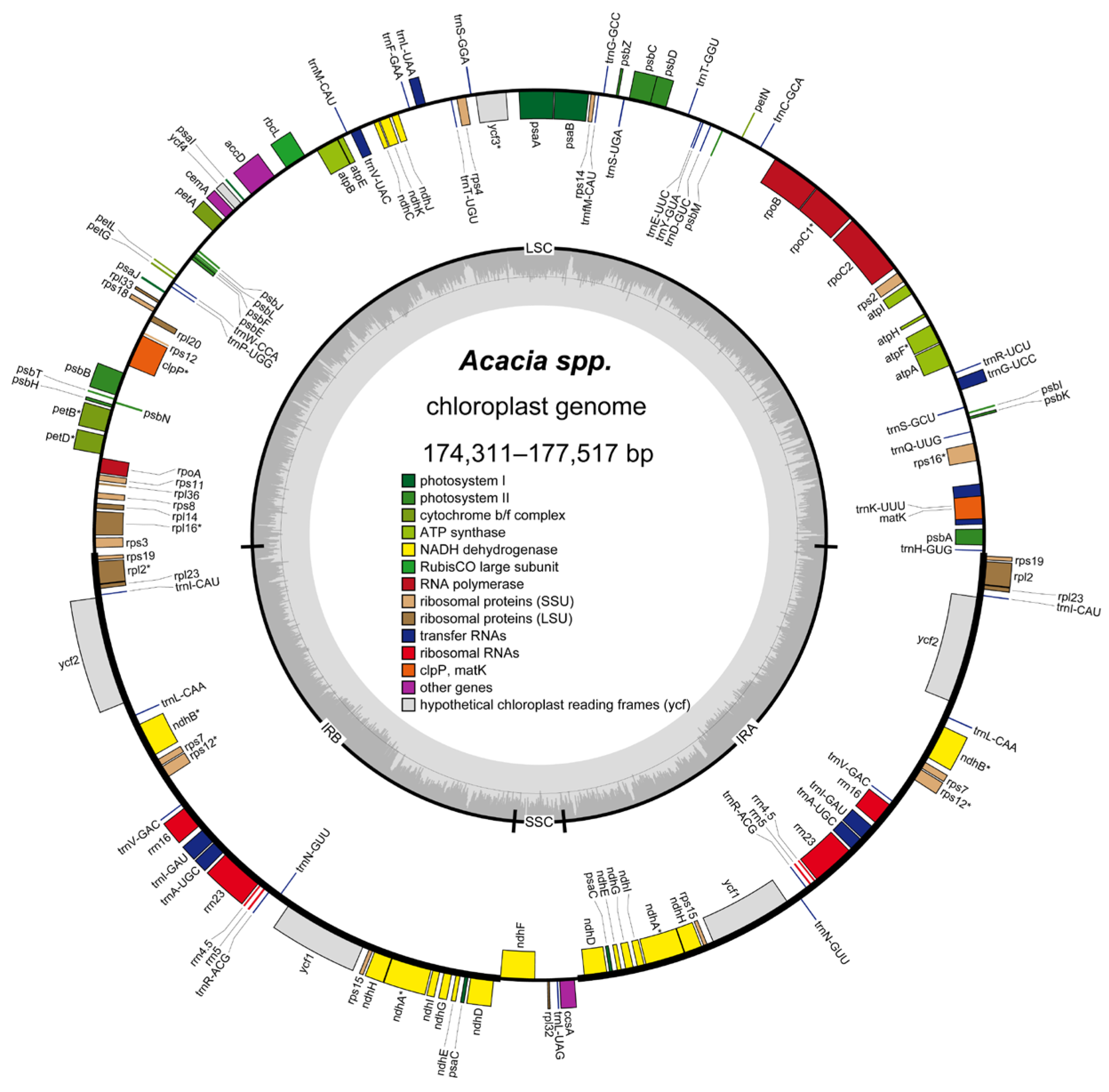
3.1. Genome Characteristics of Acacia Plastomes
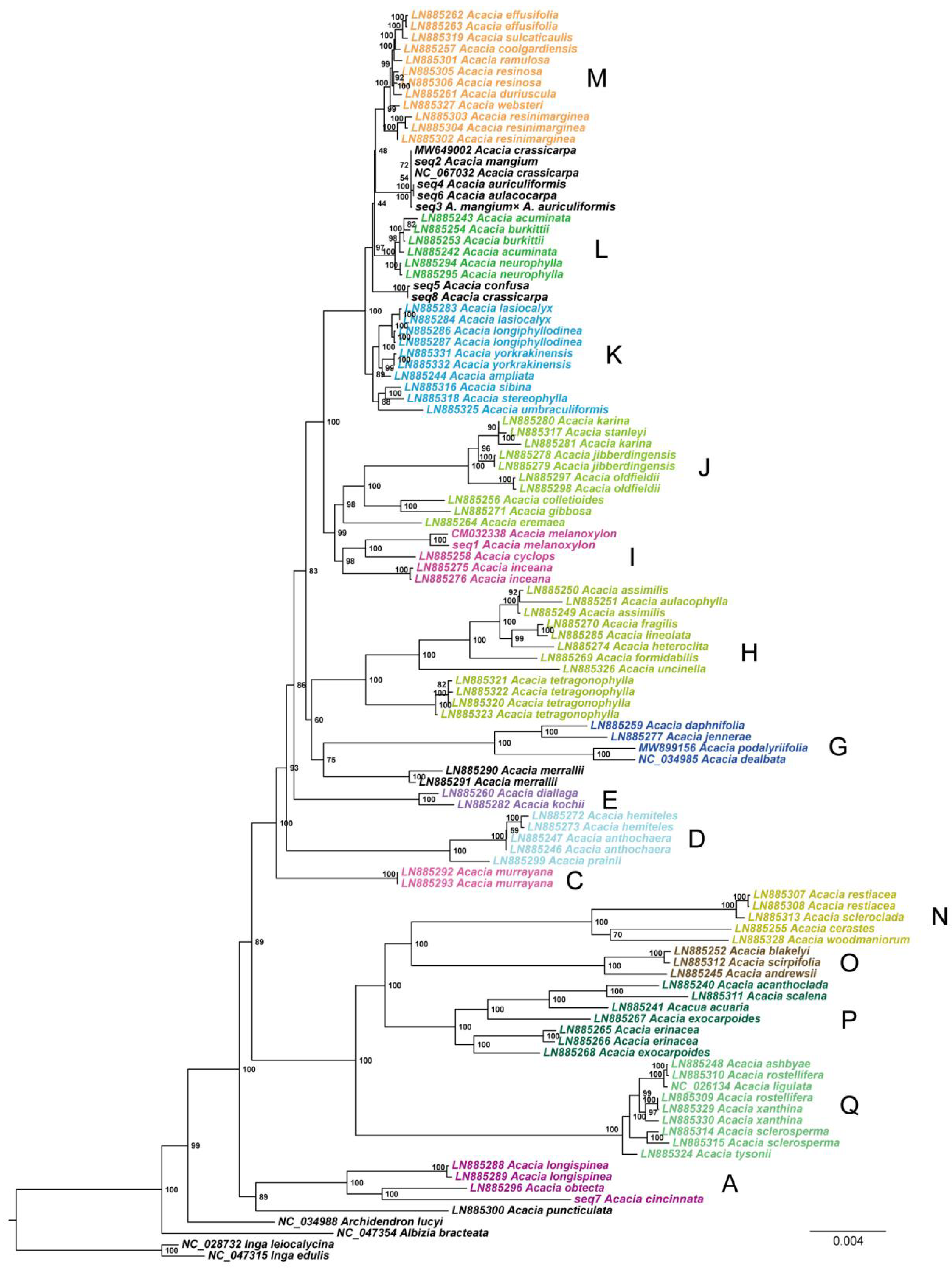
3.2. Phylogenetic Analyses
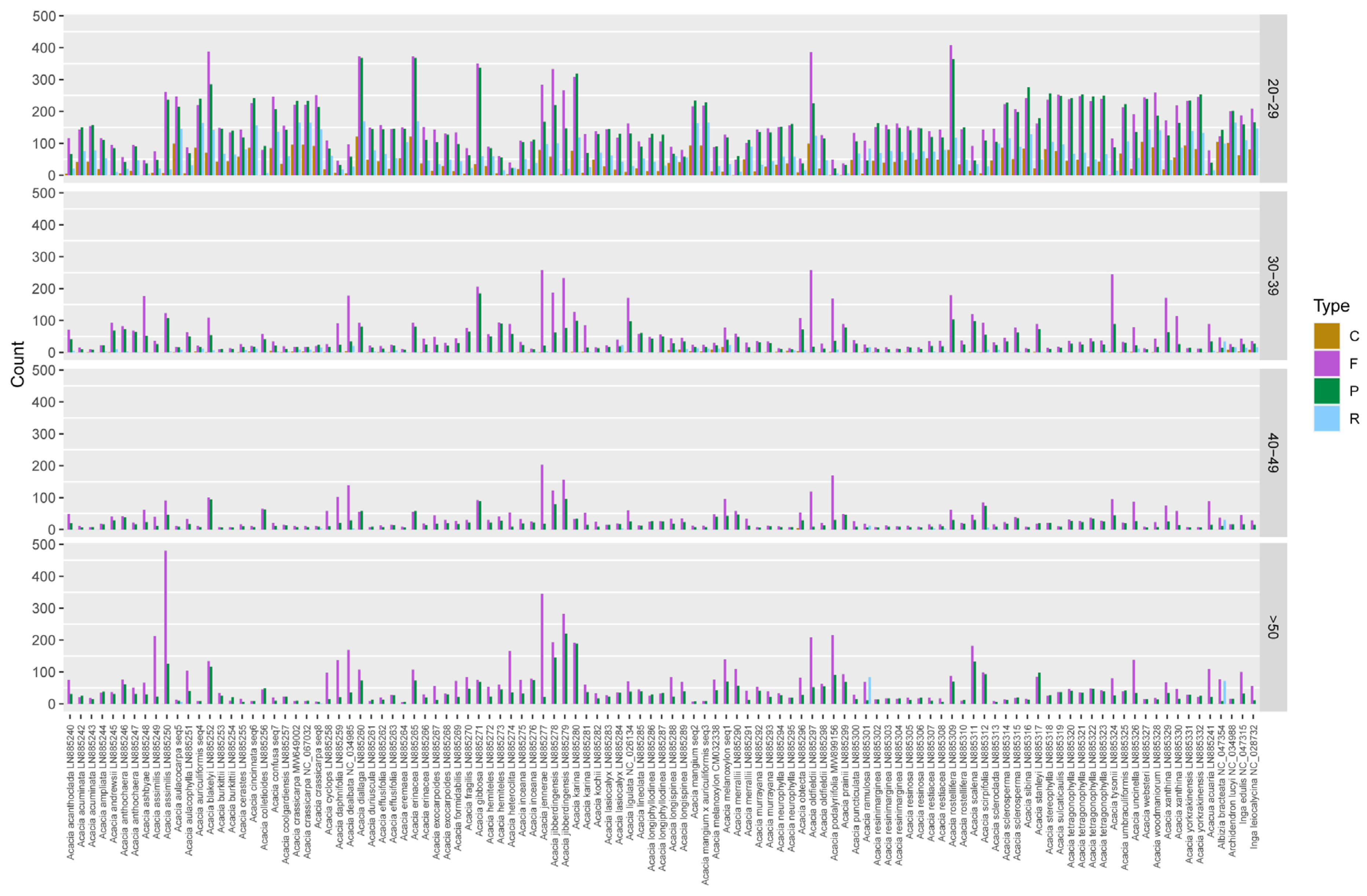
3.3. Repeat Analysis
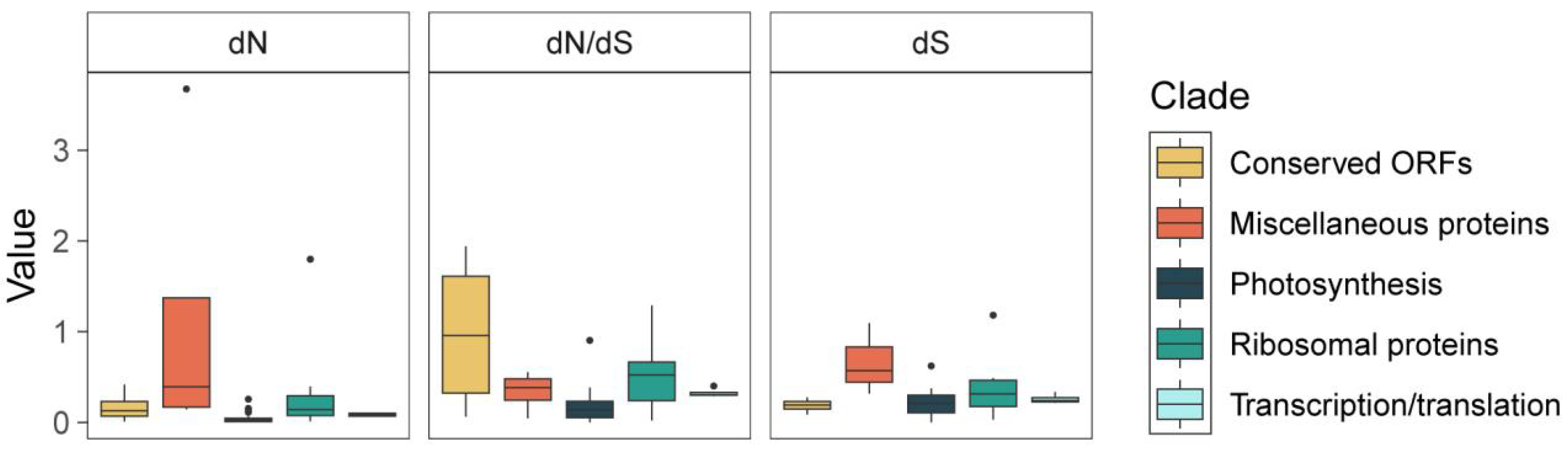
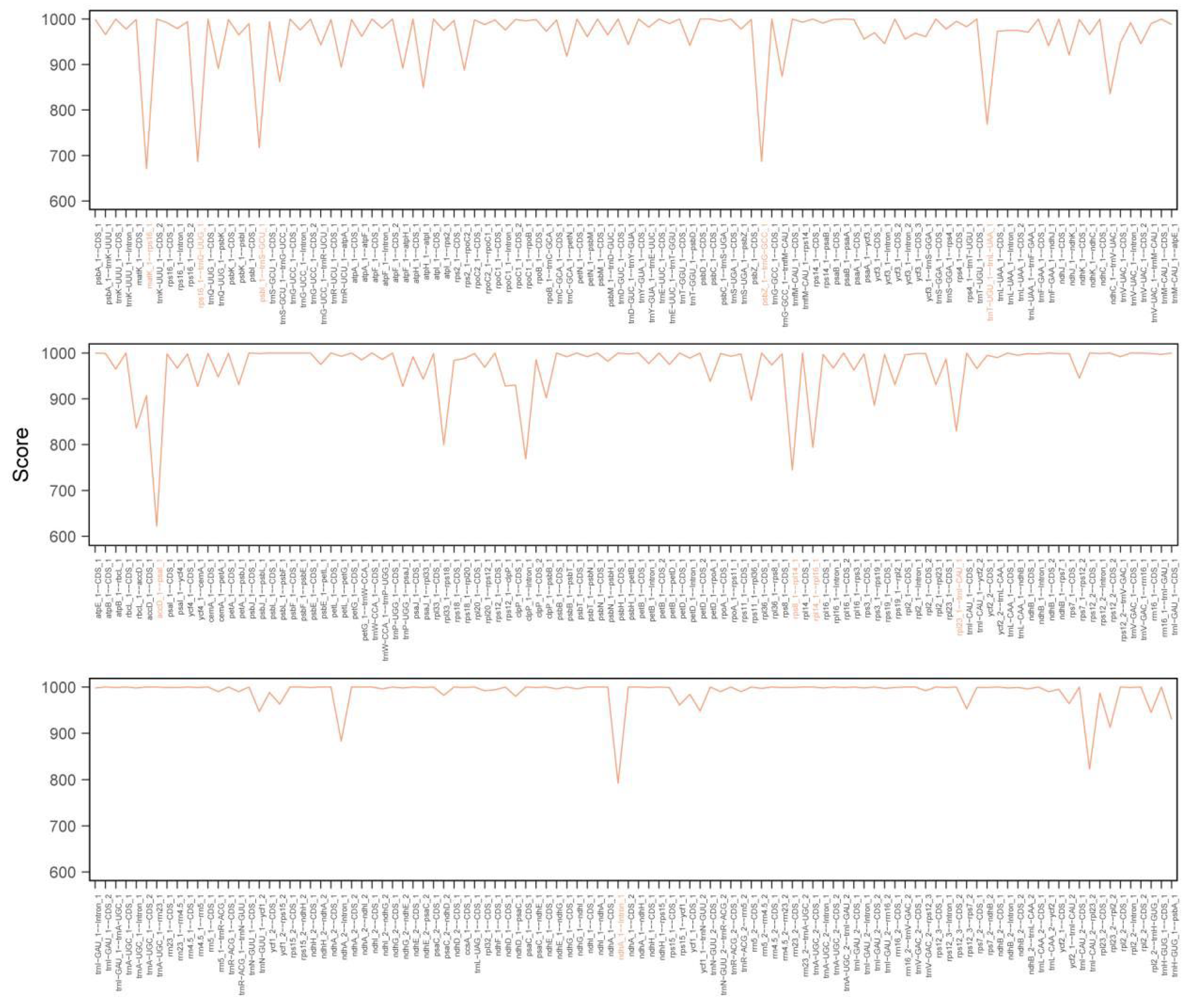
3.4. Genome Sequence Divergence and Barcode Selection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boland, D.J.; Brooker, M.I.H.; Chippendale, G.M.; Hall, N.; Hyland, B.P.M.; Johnston, R.D.; Kleinig, D.A.; McDonald, M.W.; Turner, J.D. Forest Trees of Australia, 4th ed.; CSIRO Agriculture & Food: Perth, Australia, 1984; pp. 147–167. [Google Scholar]
- Thiele, K.R.; Funk, V.A.; Iwatsuki, K.; Morat, P.; Peng, C.-I.; Raven, P.H.; Sarukhán, J.; Seberg, O. The controversy over the retypification of Acacia Mill. with an Australian type: A pragmatic view. Taxon 2011, 60, 194–198. [Google Scholar] [CrossRef]
- Chou, C.-H.; Fu, C.-Y.; Li, S.-Y.; Wang, Y.-F. Allelopathic Potential of Acacia confusa and Related Species in Taiwan. J. Chem. Ecol. 1998, 24, 2131–2150. [Google Scholar] [CrossRef]
- Luo, Y.; Li, D. Flora of China. J. Plant Classif. Resour. 2013, 6, 742810. [Google Scholar]
- Zhang, N. General review on the research development of Acacia spp. in China. Trop. For. 2015, 43, 41–45. [Google Scholar]
- Yang, M.Q. The Study on the Growth and Adaptability of Major Tropical Acacia in South China. For. Res. 1990, 3, 155–166. [Google Scholar]
- Cai, J.B.; Sun, Z.B.; Lu, R.X. Study on Drying Characteristic and Drying Technology for Rich Acaica Wood. China Wood Ind. 2002, 16, 25–30. [Google Scholar]
- Bradbury, G.; Potts, B.; Beadle, C. Genetic and environmental variation in wood properties of Acacia melanoxylon. Ann. For. Sci. 2011, 68, 1363–1373. [Google Scholar] [CrossRef]
- Nanjing, C. Kraft Pulping Properties Of Acacia Spp. Introduced From Australia. Chem. Ind. For. Prod. 1995, 15, 50–56. [Google Scholar]
- Kha L, D. The role of Acacia hybrids in the reforestation program in Vietnam. NFT News 2000, 3, 2. [Google Scholar]
- Huang, B.L. Primary Studies on Ndophytic Bacteria Isolated from Root Nodules of Acacia Confuse in Limestone Area. Master’s Thesis, Guangxi University, Nanning, China, 2009. [Google Scholar]
- Pregitzer, K.; Kubiske, M.; Yu, C.; Hendrick, R. Relationships among root branch order, carbon, and nitrogen in four temperate species. Oecologia 1997, 111, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024. [Google Scholar] [CrossRef] [PubMed]
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J. Chloroplast DNA exists in two orientations. Nature 1983, 301, 92–93. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; de Pamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [CrossRef]
- Wu, Z.; Tembrock, L.R.; Ge, S. Are differences in genomic data sets due to true biological variants or errors in genome assembly: An example from two chloroplast genomes. PLoS ONE 2015, 10, e0118019. [Google Scholar] [CrossRef]
- Xue, S.; Shi, T.; Luo, W.; Ni, X.; Iqbal, S.; Ni, Z.; Huang, X.; Yao, D.; Shen, Z.; Gao, Z. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 2019, 6, 89. [Google Scholar] [CrossRef]
- Abreu, N.; Alves, R.; Cardoso, S.; Yann, B.; Sousa, F.; Hall, C.; Pfeil, B.; Antonelli, A. The use of chloroplast genome sequences to solve phylogenetic incongruences in Polystachya Hook (Orchidaceae Juss). PeerJ 2018, 6, e4916. [Google Scholar] [CrossRef]
- Li, H.T.; Yi, T.S.; Gao, L.M.; Ma, P.F.; Zhang, T.; Yang, J.B.; Gitzendanner, M.A.; Fritsch, P.W.; Cai, J.; Luo, Y.; et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 2019, 5, 461–470. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.-F.; Yu, Y.; Deng, Y.-Q.; Li, J.; Liu, H.-Y.; Zhou, S.-D.; He, X.-J. Comparative Analysis of the Chloroplast Genomes of the Chinese Endemic Genus Urophysa and Their Contribution to Chloroplast Phylogeny and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Li, W.; Jin, X.; Hj, Z. A New Nuclear DNA Marker Revealing Both Microsatellite Variations and Single Nucleotide Polymorphic Loci: A Case Study on Classification of Cultivars in Lagerstroemia indica L. J. Microb. Biochem. Technol. 2016, 8, 266–271. [Google Scholar] [CrossRef]
- Wang, J.; Liao, X.; Gu, C.; Xiang, K.; Li, S.; Tembrock, L.R.; Wu, Z.; He, W. The Asian lotus (Nelumbo nucifera) pan-plastome: Diversity and divergence in a living fossil grown for seed, rhizome, and aesthetics. Ornam. Plant Res. 2022, 2, 1–10. [Google Scholar] [CrossRef]
- Wang, J.; Liao, X.; Li, Y.; Ye, Y.; Xing, G.; Kan, S.; Nie, L.; Li, S.; Tembrock, L.R.; Wu, Z. Comparative Plastomes of Curcuma alismatifolia (Zingiberaceae) Reveal Diversified Patterns among 56 Different Cut-Flower Cultivars. Genes 2023, 14, 1743. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Qu, X.-J.; Moore, M.J.; Li, D.-Z.; Yi, T.-S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Liao, X.; Ye, Y.; Zhang, X.; Peng, D.; Hou, M.; Fu, G.; Tan, J.; Zhao, J.; Jiang, R.; Xu, Y.; et al. The genomic and bulked segregant analysis of Curcuma alismatifolia revealed its diverse bract pigmentation. aBIOTECH 2022, 3, 178–196. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Liao, X.; Ye, Y.; Zhang, N.; Yang, Z.; Zhu, W.; Gao, W.; Sharbrough, J.; Tembrock, L.R.; Xu, D.; et al. A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genom. 2021, 22, 672. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.Z.; Liu, Y.L.; Zhang, D.; Li, W.; Gao, J.; Liu, Y.; Li, K.; Shi, C.; Zhao, Y.; Zhao, Y.J.; et al. Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2019, 2, 278. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Landis, J.B.; Lv, Z.; Shen, J.; Zhang, H.; Lin, N.; Li, L.; Sun, J.; Deng, T.; et al. Plastome phylogenomic study of Gentianeae (Gentianaceae): Widespread gene tree discordance and its association with evolutionary rate heterogeneity of plastid genes. BMC Plant Biol. 2020, 20, 340. [Google Scholar] [CrossRef] [PubMed]
- Mishler, B.D.; Knerr, N.; Gonzalez-Orozco, C.E.; Thornhill, A.H.; Laffan, S.W.; Miller, J.T. Phylogenetic measures of biodiversity and neo- and paleo-endemism in Australian Acacia. Nat. Commun. 2014, 5, 4473. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.V.; Miller, J.T.; Small, I.; Nevill, P.G.; Boykin, L.M. Integration of complete chloroplast genome sequences with small amplicon datasets improves phylogenetic resolution in Acacia. Mol. Phylogenet. Evol. 2016, 96, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brázda, V.; Lýsek, J.; Bartas, M.; Fojta, M. Complex Analyses of Short Inverted Repeats in All Sequenced Chloroplast DNAs. Biomed Res. Int. 2018, 2018, 1097018. [Google Scholar] [CrossRef] [PubMed]
- Saltonstall, K.; Lambertini, C. The value of repetitive sequences in chloroplast DNA for phylogeographic inference: A comment on Vachon & Freeland 2011. Mol. Ecol. Resour. 2012, 12, 581–585, discussion 586–589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.R.; Yang, X.; Li, W.Y.; Peng, Y.Q.; Gao, J. Comparative chloroplast genome analysis of Ficus (Moraceae): Insight into adaptive evolution and mutational hotspot regions. Front. Plant Sci. 2022, 13, 965335. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GB ID | Species | TL | Genes | tRNA | rRNA | GC | LSC | IRB | SSC | IRA |
|---|---|---|---|---|---|---|---|---|---|---|
| seq1 | Acacia melanoxylon | 177,517 | 77 | 30 | 4 | 35.33% | 90,996 | 40,813 | 4895 | 40,813 |
| seq2 | Acacia mangium | 176,085 | 77 | 30 | 4 | 35.37% | 92,172 | 39,482 | 4949 | 39,482 |
| seq3 | A. mangium × A. auriculiformis | 176,658 | 77 | 30 | 4 | 35.32% | 91,991 | 39,859 | 4949 | 39,859 |
| seq4 | Acacia auriculiformis | 176,282 | 77 | 30 | 4 | 35.38% | 91,638 | 39,859 | 4926 | 39,859 |
| seq5 | Acacia confusa | 176,255 | 77 | 30 | 4 | 35.45% | 90,722 | 40,243 | 5047 | 40,243 |
| seq6 | Acacia aulacocarpa | 176,515 | 77 | 30 | 4 | 35.34% | 91,871 | 39,859 | 4926 | 39,859 |
| seq7 | Acacia cincinnata | 174,311 | 77 | 30 | 4 | 35.66% | 90,325 | 39,560 | 4866 | 39,560 |
| seq8 | Acacia crassicarpa | 176,659 | 77 | 30 | 4 | 35.38% | 91,220 | 40,197 | 5045 | 40,197 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Li, Y.; Tembrock, L.R.; Wu, Z.; Liao, X.; Zhang, S. Comparative Analyses of Acacia Plastomes to Detect Mutational Hotspots and Barcode Sites for the Identification of Important Timber Species. Forests 2024, 15, 306. https://doi.org/10.3390/f15020306
Li W, Li Y, Tembrock LR, Wu Z, Liao X, Zhang S. Comparative Analyses of Acacia Plastomes to Detect Mutational Hotspots and Barcode Sites for the Identification of Important Timber Species. Forests. 2024; 15(2):306. https://doi.org/10.3390/f15020306
Chicago/Turabian StyleLi, Weixiong, Yongyao Li, Luke R. Tembrock, Zhiqiang Wu, Xuezhu Liao, and Shuo Zhang. 2024. "Comparative Analyses of Acacia Plastomes to Detect Mutational Hotspots and Barcode Sites for the Identification of Important Timber Species" Forests 15, no. 2: 306. https://doi.org/10.3390/f15020306
APA StyleLi, W., Li, Y., Tembrock, L. R., Wu, Z., Liao, X., & Zhang, S. (2024). Comparative Analyses of Acacia Plastomes to Detect Mutational Hotspots and Barcode Sites for the Identification of Important Timber Species. Forests, 15(2), 306. https://doi.org/10.3390/f15020306