Combined Analysis of mRNAs and miRNAs to Identify Genes Related to Biological Characteristics of Autotetraploid Paulownia

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA Isolation
2.2. Construction of the Paulownia cDNA Libraries and Transcriptome Sequencing Data Processing
2.3. Construction of Paulownia Small RNA Libraries and Small RNA Sequencing Data Processing
counts in the diploid library)
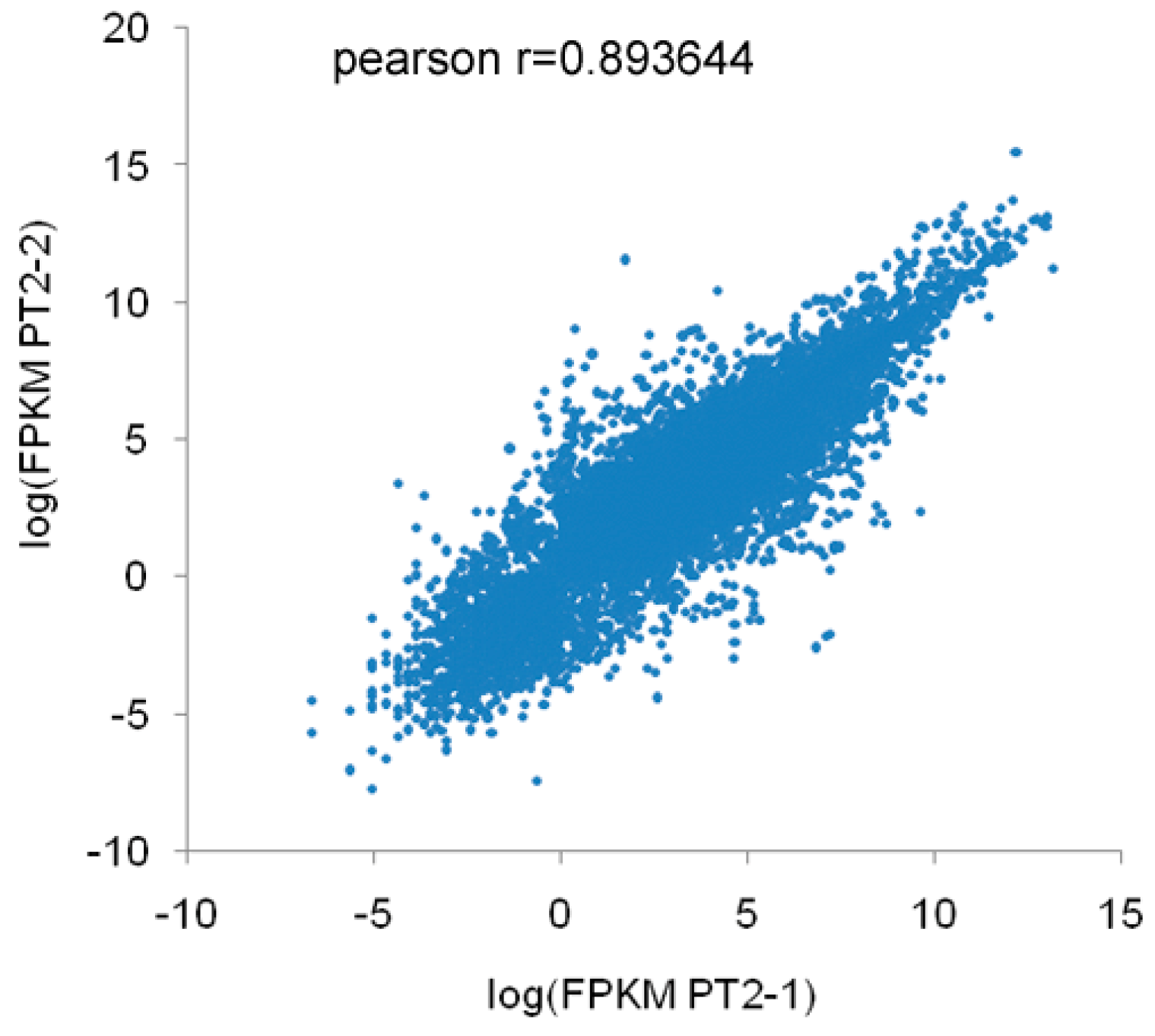
2.4. Correlation Analysis of mRNA and miRNA
2.5. Verification of DEGs and DEMs by Quantitative Real-Time Polymerase Chain Reaction
3. Results
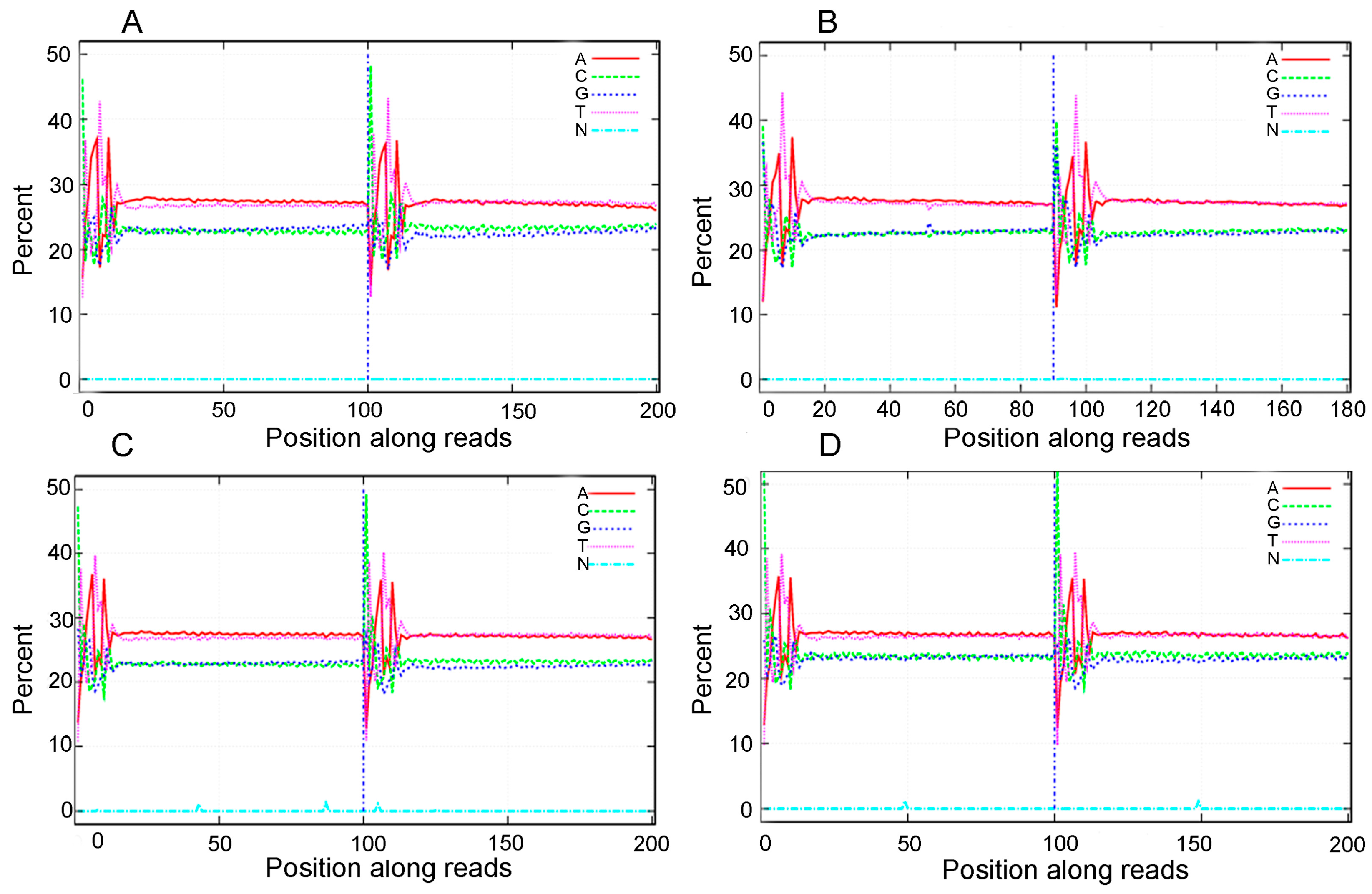
3.1. Analysis of the Transcriptome Sequencing Data
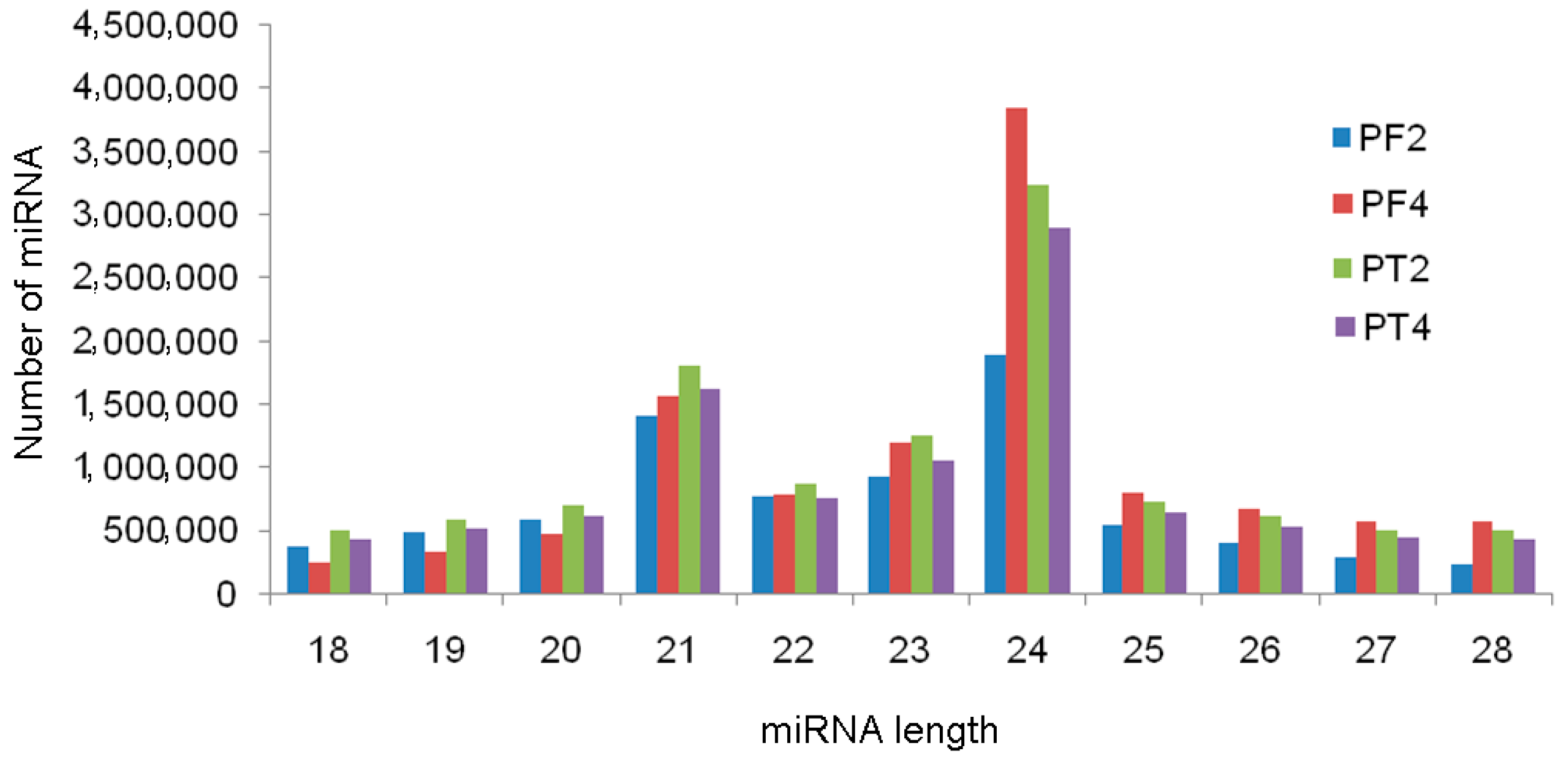
3.2. Analysis of the miRNA Sequencing Data
3.3. Identification of the Key Genes Related to the Biological Characteristics of Tetraploid Paulownia
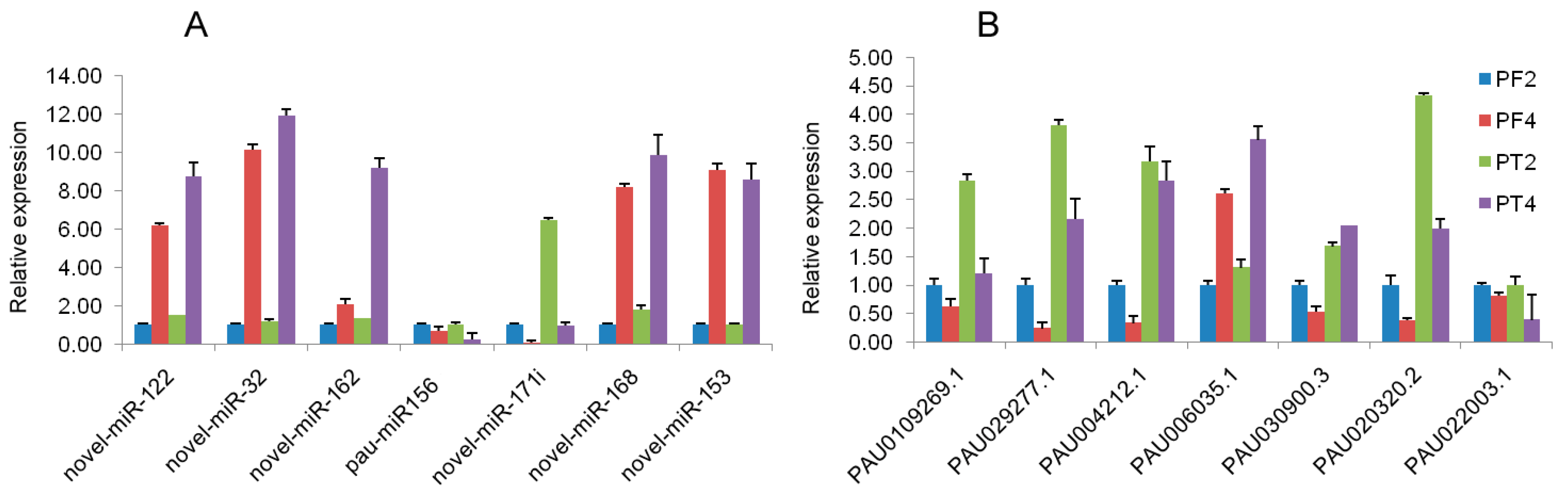
3.4. qRT-PCR Validation
4. Discussion
4.1. DEMs Mediated Their Target Genes That Increased the Abiotic Stress Tolerance in Tetraploid Paulownia
4.2. DEM Target Gene Involved in Epigenetic Alterations That Regulated Tetraploid Paulownia Timber Quality
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dhooghe, E.; Van Laere, K.; Eeckhaut, T.; Leus, L.; Van Huylenbroeck, J. Mitotic chromosome doubling of plant tissues in vitro. Plant Cell Tissue Org. Cult. 2011, 104, 359–373. [Google Scholar] [CrossRef]
- Doumett, S.; Lamperi, L.; Checchini, L.; Azzarello, E.; Mugnai, S.; Mancuso, S.; Petruzzelli, G.; Del Bubba, M. Heavy metal distribution between contaminated soil and Paulownia tomentosa, in a pilot-scale assisted phytoremediation study: Influence of different complexing agents. Chemosphere 2008, 72, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Podda, A.; Checcucci, G.; Mouhaya, W.; Centeno, D.; Rofidal, V.; Del Carratore, R.; Luro, F.; Morillon, R.; Ollitrault, P.; Maserti, B.E. Salt-stress induced changes in the leaf proteome of diploid and tetraploid mandarins with contrasting Na+ and CL− accumulation behaviour. J. Plant Physiol. 2013, 170, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Wang, Z.; Luo, G.; Tang, C. Phenotypic and transcriptomic analyses of autotetraploid and diploid mulberry (Morus alba L.). Int. J. Mol. Sci. 2015, 16, 22938–22956. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.H.; Liu, P.; Liu, M.J.; Wang, J.R.; Xu, J. A novel method for rapid in vivo induction of homogeneous polyploids via calluses in a woody fruit tree (Ziziphus jujuba Mill.). Plant Cell Tissue Org. Cult. 2015, 121, 423–433. [Google Scholar] [CrossRef]
- Dudits, D.; Török, K.; Cseri, A.; Paul, K.; Nagy, A.V.; Nagy, B.; Sass, L.; Ferenc, G.; Vankova, R.; Dobrev, P. Response of organ structure and physiology to autotetraploidization in early development of energy willow Salix viminalis. Plant Physiol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krikorian, A.D. Paulownia in china: Cultivation and utilization. Econ. Bot. 1988, 42, 283. [Google Scholar] [CrossRef]
- Jey, A. Paulownia Plantation Experiences and Profitable Timber Production. In Proceedings of the Plantation and Regrowth Forestry—A diversity of opportunity, Australian Forest Growers Biennial Conference, Lismore, Australia, 6–9 July 1998; pp. 199–214. [Google Scholar]
- Kalaycioglu, H.; Deniz, I.; Hiziroglu, S. Some of the properties of particleboard made from paulownia. J. Wood Sci. 2005, 51, 410–414. [Google Scholar] [CrossRef]
- Ipekci, Z.; Gozukirmizi, N. Direct somatic embryogenesis and synthetic seed production from Paulownia elongata. Plant Cell Rep. 2003, 22, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Yang, Z.; Cao, Y.; Liu, F.; Jia, F. Autotetraploid induction of Paulownia elongata with colchione. J. Nucl. Agric. Sci. 2006, 20, 473–476. [Google Scholar]
- Fan, G.; Zhai, X.; Wei, Z.; Yang, Z. Induction of autotrtraploid from the saomtic cell of Paulownia tomentosa × Paulownia fortunei and its in vitro plantlet regeneration. J. NE Forestry. Univ. 2010, 38, 22–26. [Google Scholar]
- Fan, G.; Wei, Z.; Yang, Z. Induction of autotetraploid of Paulownia australis and its in vitro plantlet regeneration. J. Northwest A F Univ. 2009, 37, 83–90. [Google Scholar]
- Fan, G.; Zhang, X.; Deng, M. Induction of autotetraploid Paulownia fortunei. Sci. Silv. Sin. 2007, 43, 31–35. [Google Scholar]
- Fan, G.Q.; Cao, Y.C.; Zhai, Z.; Yang, Z.Q. Induction of autotetraploid of Paulownia tomentosa (Thunb.) steud. Plant Physiol. Commun. 2007, 109, 43. [Google Scholar]
- Zhai, X.; Zhang, X.; Zhao, Z.; Deng, M.; Fan, G. Study on wood physical properties of tetraploid Paulownia fortunei. J. Henan Agric. Univ. 2012, 46, 651–654. [Google Scholar]
- Zhang, X.; Deng, M.; Fan, G. Differential transcriptome analysis between Paulownia fortunei and its synthesized autopolyploid. Int. J. Mol. Sci. 2014, 15, 5079–5093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fan, G.; Zhao, Z.; Cao, X.; Zhao, G.; Deng, M.; Dong, Y. Analysis of diploid and its autotetraploid Paulownia tomentosa × Paulownia fortunei with AFLP and MSAP. Sci. Silv. Sin. 2013, 49, 167–172. [Google Scholar]
- Zhai, X.; Zhang, X.; Fan, G.; Zhao, Z.; Cao, X. Analysis of diploid and its autotetraploid Paulownia tomentosa with AFLP and MSAP. J. Henan Agric. Univ. 2015, 49, 89–93. [Google Scholar]
- Deng, M.; Zhang, X.; Fan, G.; Zhao, Z.; Dong, Y.; Wei, Z. Comparative studies on physiological responses to salt stress in tetraploid paulownia plants. J. Cent. South Univ. For. Technol. 2013, 33, 42–46. [Google Scholar]
- Paulownia Genome Database, Henan Agricultural University. Available online: http://paulownia.genomics.cn (accessed on 21 March 2013).
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 2010, 15, 473–497. [Google Scholar] [CrossRef]
- Fan, G.; Wang, L.; Dong, Y.; Zhao, Z.; Deng, M.; Niu, S.; Zhang, X.; Cao, X. Genome of paulownia (Paulownia fortunei) illuminates the related transcripts, mirRNA and proteins for salt resistance. Sci. Rep. 2017, 7, 1285. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.K. Te expectation-maximization algorithm. IEEE Signal Process. Mag. 2000, 13, 47–60. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from rna-seq data with or without a reference genome. BMC Bioinform. 2011. [Google Scholar] [CrossRef] [PubMed]
- Broberg, P. A comparative review of estimates of the proportion unchanged genes and the false discovery rate. BMC Bioinform. 2005, 6, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Fan, G.; Zhai, X.; Niu, S.; Ren, Y. Dynamic expression of novel and conserved microRNAs and their targets in diploid and tetraploid of Paulownia tomentosa. Biochimie 2014, 102, 68–77. [Google Scholar] [CrossRef] [PubMed]
- BLAST. Available online: http: //www.ncbi.nlm.nih.gov/ (accessed on 29 January 2015).
- Rfam. Available online: http://rfam.sanger.ac.uk/ (accessed on 11 November 2014).
- MicroRNA Discovery By Deep Sequencing. Available online: http://sourceforge.net/projects/mireap/ (accessed on 27 March 2013).
- RNAfold web server. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi (accessed on 10 January 1981).
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- carringtonlab/TargetFinder. Available online: https://github.com/carringtonlab/TargetFinder (accessed on 17 March 2014).
- psRobot: Plant Small RNA Analysis Toolbox. Available online: http://omicslab.genetics.ac.cn/psRobot (accessed on 12 March 2012).
- Saminathan, T.; Nimmakayala, P.; Manohar, S.; Malkaram, S.; Almeida, A.; Cantrell, R.; Tomason, Y.; Abburi, L.; Rahman, M.A.; Vajja, V.G. Differential gene expression and alternative splicing between diploid and tetraploid watermelon. J. Exp. Bot. 2014, 66, 1369–1385. [Google Scholar] [CrossRef] [PubMed]
- Podwyszyńska, M.; Gabryszewska, E.; Dyki, B.; Stępowska, A.; Kowalski, A.; Jasiński, A. Phenotypic and genome size changes (variation) in synthetic tetraploids of daylily (Hemerocallis) in relation to their diploid counterparts. Euphytica 2015, 203, 1–16. [Google Scholar] [CrossRef]
- Balal, R.M.; Shahid, M.A.; Vincent, C.; Zotarelli, L.; Liu, G.; Mattson, N.S.; Rathinasabapathi, B.; Martínez-Nicolas, J.J.; Garcia-Sanchez, F. Kinnow mandarin plants grafted on tetraploid rootstocks are more tolerant to cr-toxicity than those grafted on its diploids one. Environ. Exp. Bot. 2017, 140, 8–18. [Google Scholar] [CrossRef]
- Niu, S.; Fan, G.; Zhao, Z.; Deng, M.; Dong, Y. High-throughput sequencing and degradome analysis reveal microRNA differential expression profiles and their targets in Paulownia fortunei. Plant Cell Tissue Org. Cult. 2014, 119, 457–468. [Google Scholar] [CrossRef]
- Fan, G.; Wang, L.; Deng, M.; Niu, S.; Zhao, Z.; Xu, E.; Cao, X.; Zhang, X. Transcriptome analysis of the variations between autotetraploid Paulownia tomentosa and its diploid using high-throughput sequencing. Mol. Genet. Genom. 2015, 290, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, J.; Sanchez-Ferrero, J.C.; Langridge, P.; Milne, L.; Chowdhury, J.; Brien, C.; Tricker, P.J. Differential expression of micrornas and potential targets under drought stress in barley. Plant Cell Environ. 2017, 40, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Ma, Y.; Wang, X.F.; Zhang, D.P. Overexpression of the transcription factor NF-YC9 confers abscisic acid hypersensitivity in Arabidopsis. Plant Mol. Biol. 2017, 95, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Myers, Z.A.; Kumimoto, R.W.; Siriwardana, C.L.; Gayler, K.K.; Risinger, J.R.; Pezzetta, D.; Holt III, B.F. Nuclear factor Y, subunit C (NF-YC) transcription factors are positive regulators of photomorphogenesis in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006333. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Qiao, Y.; Mao, K.; Hao, Y.; You, C. Overexpression of arabidopsis atmiR408 gene in tobacco. Acta Biol. Crac. Bot. 2010, 52, 26–31. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, Y.; Zhuo, C.; Lu, S.; Guo, Z. Overexpression of a NF-YC transcription factor from bermudagrass confers tolerance to drought and salinity in transgenic rice. Plant Biotechnol. J. 2015, 13, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Verbinnen, I.; Ferreira, M.; Bollen, M. Biogenesis and activity regulation of protein phosphatase 1. Biochem. Soc. Trans. 2017, 45, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.D.; Lin, K.H.; Chen, C.C.; Chiang, C.M. Oryza sativa protein phosphatase 1a (OsPP1a) involved in salt stress tolerance in transgenic rice. Mol. Breed. 2016, 36, 1–19. [Google Scholar] [CrossRef]
- Wang, L.; Mai, Y.X.; Zhang, Y.C.; Luo, Q.; Yang, H.Q. MicroRNA171c-targeted SCL6-II, SCL6-III, and SCL6-IV genes regulate shoot branching in Arabidopsis. Mol. Plant 2010, 3, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Takuno, S.; Ran, J.; Gaut, B.S. Evolutionary patterns of genic DNA methylation vary across land plants. Nat. Plants 2016. [Google Scholar] [CrossRef] [PubMed]
- Lang, Z.; Xie, S.; Zhu, J.-K. The 1001 arabidopsis DNA methylomes: An important resource for studying natural genetic, epigenetic, and phenotypic variation. Trends Plant Sci. 2016, 21, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Xia, E.; Yao, Q.; Liu, X.; Gao, L. Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, E7022–E7029. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, L.-J.; Quinton-Tulloch, M.; Olohan, L.; Price, J.; Hall, N.; Hall, A. A genome-wide survey of DNA methylation in hexaploid wheat. Genome Biol. 2015, 16, 273. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Wang, R.; Wang, J. Correlation analysis of the mRNA and miRNA expression profiles in the nascent synthetic allotetraploid Raphanobrassica. Sci. Rep. 2016, 6, 37416. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef] [PubMed]
- Lewsey, M.G.; Hardcastle, T.J.; Melnyk, C.W.; Molnar, A.; Valli, A.; Urich, M.A.; Nery, J.R.; Baulcombe, D.C.; Ecker, J.R. Mobile small rnas regulate genome-wide DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, E801–E810. [Google Scholar] [CrossRef] [PubMed]
- Kaneshiro, E.S.; Johnston, L.Q.; Nkinin, S.W.; Romero, B.I.; Giner, J.L. Sterols of saccharomyces cerevisiae erg6 knockout mutant expressing the pneumocystis carinii S-adenosylmethionine: Sterol c-24 methyltransferase. J. Eukaryot. Microbiol. 2015, 62, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gonzales, R.A.; Bhattacharyya, M.K. Identification and characterization of an S-adenosyl-L-methionine:-sterol-c-methyltransferase cDNA from soybean. J. Biol. Chem. 1996, 271, 9384–9389. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Do, J.; Shin, S.J.; Choi, J.W.; Choi, Y.I.; Kim, W.; Kwon, M. Exogenously applied 24-epi brassinolide reduces lignification and alters cell wall carbohydrate biosynthesis in the secondary xylem of liriodendron tulipifera. Phytochemistry 2014, 101, 40–51. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | GC (%) | Q20 (%) | Q30 (%) | Adapter | N | Low Quality Reads | Clean Reads | Ratio of Clean Reads |
|---|---|---|---|---|---|---|---|---|---|
| PF2 | 60,025,508 | 46.43 | 91.06 | 84.91 | 0 | 68,226 | 4,541,576 | 55,415,706 | 92.32% |
| PF4 | 63,050,814 | 45.78 | 95.65 | 90.37 | 170,350 | 278,396 | 2,417,730 | 60,184,338 | 95.45% |
| PT2 | 37,819,270 | 46.17 | 94.87 | 90.83 | 0 | 18,780 | 1,496,634 | 36,303,856 | 95.99% |
| PT4 | 37,817,466 | 47.1 | 94.67 | 90.70 | 0 | 17,904 | 1,602,808 | 36,196,754 | 95.71% |
| Sample | Total Reads | High Quality | 3′adapter Null | Insert Null | 5′adapter Contaminants | Smaller than 18 nt | polyA | Clean Reads |
|---|---|---|---|---|---|---|---|---|
| PF2 | 9,774,977 | 9,679,547 | 406,061 | 483,751 | 13,380 | 884,549 | 211 | 7,891,595 |
| 100% | 4.20% | 5.00% | 0.14% | 9.14% | 0.00% | 81.53% | ||
| PF4 | 14,422,555 | 14,176,998 | 2,149,291 | 49,377 | 14,150 | 554,768 | 435 | 11,018,977 |
| 100% | 15.16% | 3.10% | 0.10% | 3.91% | 0.00% | 77.72% | ||
| PT2 | 14,520,461 | 14,261,213 | 1,607,505 | 161,420 | 14,284 | 1,230,980 | 362 | 11,246,662 |
| 100% | 11.27% | 1.13% | 0.10% | 8.63% | 0.00% | 78.86% | ||
| PT4 | 13,109,201 | 13,008,182 | 1,407,359 | 495,415 | 23,817 | 1,196,260 | 265 | 9,885,066 |
| 100% | 10.82% | 3.81% | 0.18% | 9.20% | 0.00% | 75.99% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Xu, E.; Zhai, X.; Dong, Y.; Fan, G. Combined Analysis of mRNAs and miRNAs to Identify Genes Related to Biological Characteristics of Autotetraploid Paulownia. Forests 2017, 8, 501. https://doi.org/10.3390/f8120501
Cao X, Xu E, Zhai X, Dong Y, Fan G. Combined Analysis of mRNAs and miRNAs to Identify Genes Related to Biological Characteristics of Autotetraploid Paulownia. Forests. 2017; 8(12):501. https://doi.org/10.3390/f8120501
Chicago/Turabian StyleCao, Xibing, Enkai Xu, Xiaoqiao Zhai, Yanpeng Dong, and Guoqiang Fan. 2017. "Combined Analysis of mRNAs and miRNAs to Identify Genes Related to Biological Characteristics of Autotetraploid Paulownia" Forests 8, no. 12: 501. https://doi.org/10.3390/f8120501
APA StyleCao, X., Xu, E., Zhai, X., Dong, Y., & Fan, G. (2017). Combined Analysis of mRNAs and miRNAs to Identify Genes Related to Biological Characteristics of Autotetraploid Paulownia. Forests, 8(12), 501. https://doi.org/10.3390/f8120501