Analysis of Environment-Marker Associations in American Chestnut



Abstract
:1. Introduction
2. Materials and Methods
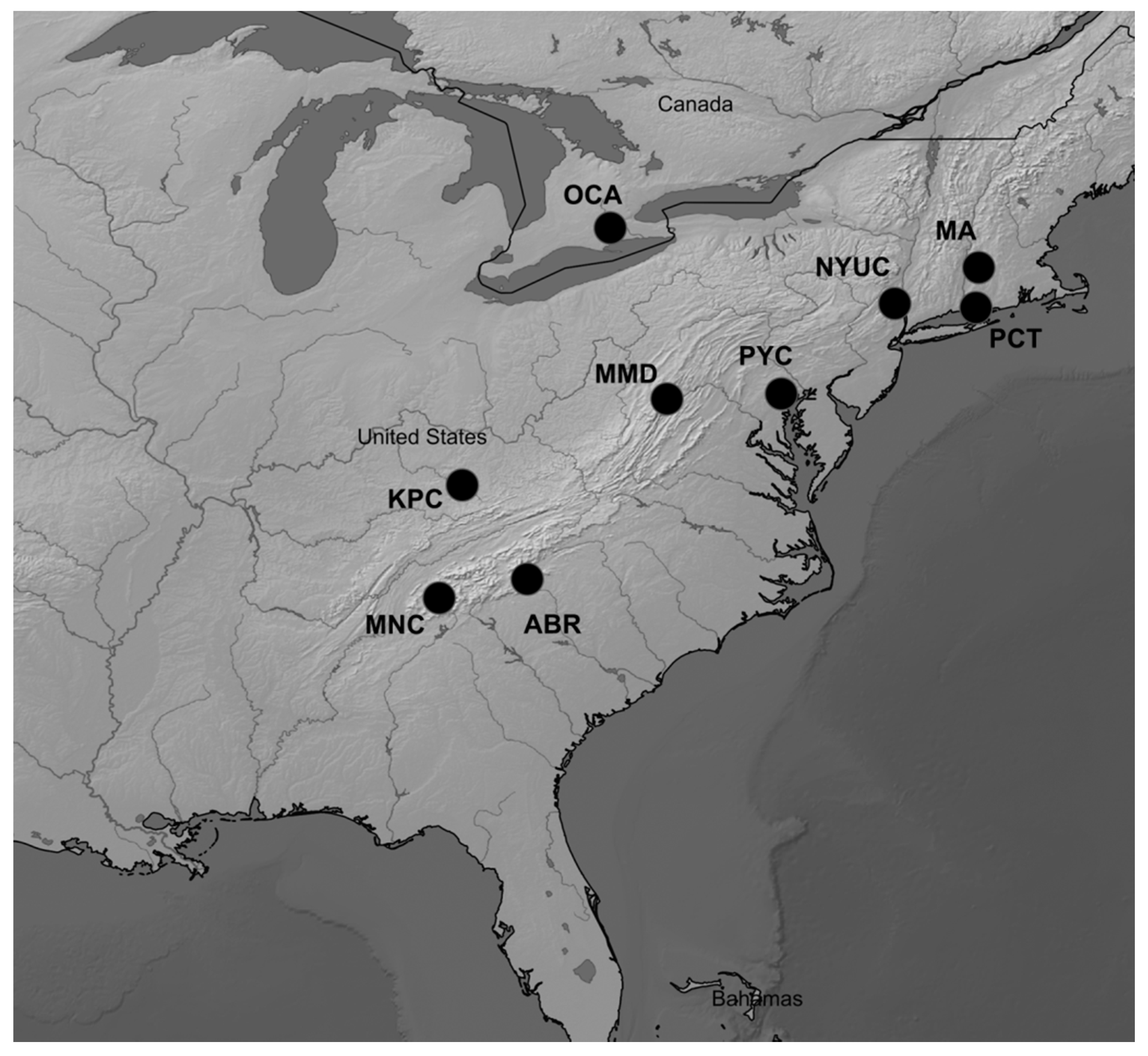
2.1. Plant Material and Environmental Variables
2.2. SSR Genotyping
2.3. Data Analysis
3. Results
3.1. Environmental Variables
3.2. Genetic Diversity and Population Structure
3.3. Outlier and Environmental Association Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jacobs, D.F.; Dalgleish, H.J.; Nelson, C.D. A conceptual framework for restoration of threatened plants: The effective model of American chestnut (Castanea dentata) reintroduction. New Phytol. 2013, 197, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Ellison, A.M.; Bank, M.S.; Clinton, B.D.; Colburn, E.A.; Elliott, K.; Ford, C.R.; Foster, D.R.; Kloeppel, B.D.; Knoepp, J.D.; Lovett, G.M.; et al. Loss of foundation species: Consequences for the structure and dynamics of forested ecosystems. Front. Ecol. Environ. 2005, 3, 479–486. [Google Scholar] [CrossRef]
- Anagnostakis, S.L. The effect of multiple importations of pests and pathogens on a native tree. Biol. Invasions 2001, 3, 245–254. [Google Scholar] [CrossRef]
- Kubisiak, T.L.; Hebard, F.V.; Nelson, C.D.; Zhang, J.; Bernatzky, R.; Huang, H.; Anagnostakis, S.L.; Doudrick, R.L. Molecular mapping of resistance to blight in an interspecific cross in the genus Castanea. Phytopathology 1997, 87, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Milgroom, M.G.; Cortesi, P. Biological control of chestnut blight with hypovirulence: A critical analysis. Annu. Rev. Phytopathol. 2004, 42, 311–338. [Google Scholar] [CrossRef] [PubMed]
- Merkle, S.A.; Andrade, G.M.; Nairn, C.J.; Powell, W.A.; Maynard, C.A. Restoration of threatened species: A noble cause for transgenic trees. Tree Genet. Genomes 2007, 3, 111–118. [Google Scholar] [CrossRef]
- Hebard, F. The backcross breeding program of the American Chestnut Foundation. J. Am. Chestnut Found. 2005, 19, 55–77. [Google Scholar]
- Bauman, J.M.; Keiffer, C.H.; McCarthy, B.C. Growth performance and chestnut blight incidence (Cryphonectria parasitica) of backcrossed chestnut seedlings in surface mine restoration. New For. 2014, 45, 813–828. [Google Scholar] [CrossRef]
- Clark, S.L.; Schlarbaum, S.E.; Saxton, A.M.; Hebard, F.V. Establishment of American chestnuts (Castanea dentata) bred for blight (Cryphonectria parasitica) resistance: Influence of breeding and nursery grading. New For. 2016, 47, 243–270. [Google Scholar] [CrossRef]
- Barakat, A.; Staton, M.; Cheng, C.H.; Park, J.; Yassin, N.B.; Ficklin, S.; Yeh, C.C.; Hebard, F.; Baier, K.; Powell, W.; et al. Chestnut resistance to the blight disease: Insights from transcriptome analysis. BMC Plant Biol. 2012, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Kubisiak, T.L.; Nelson, C.D.; Staton, M.E.; Zhebentyayeva, T.; Smith, C.; Olukolu, B.A.; Fang, G.C.; Hebard, F.V.; Anagnostakis, S.; Wheeler, N.; et al. A transcriptome-based genetic map of Chinese chestnut (Castanea mollissima) and identification of regions of segmental homology with peach (Prunus persica). Tree Genet. Genomes 2013, 9, 557–571. [Google Scholar] [CrossRef]
- Bodénès, C.; Chancerel, E.; Gailing, O.; Vendramin, G.G.; Bagnoli, F.; Durand, J.; Goicoechea, P.G.; Soliani, C.; Villani, F.; Mattioni, C.; et al. Comparative mapping in the Fagaceae and beyond with EST-SSRs. BMC Plant Biol. 2012, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Staton, M.; Zhebentyayeva, T.; Olukolu, B.; Fang, G.C.; Nelson, D.; Carlson, J.E.; Abbott, A.G. Substantial genome synteny preservation among woody angiosperm species: Comparative genomics of Chinese chestnut (Castanea mollissima) and plant reference genomes. BMC Genom. 2015, 16, 744. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, W.; Lan, Y.; Cao, Q.; Su, S.; Zhou, Z.; Wang, J.; Liu, J.; Hu, G. The complete chloroplast genome sequence of the wild Chinese chestnut (Castanea mollissima). Conserv. Genet. Resour. 2017. [Google Scholar] [CrossRef]
- Gurney, K.M.; Schaberg, P.G.; Hawley, G.J.; Shane, J.B. Inadequate cold tolerance as a possible limitation to American chestnut restoration in the Northeastern United States. Restor. Ecol. 2011, 19, 55–63. [Google Scholar] [CrossRef]
- Gailing, O.; Nelson, C.D. Genetic variation patterns of American chestnut populations at EST-SSRs. Botany 2017, 95, 799–807. [Google Scholar] [CrossRef]
- Kubisiak, T.L.; Roberds, J.H. Genetic Structure of American Chestnut Populations Based on Neutral DNA Markers; U.S. Department of the Interior, National Park Service, National Capital Region, Center for Urban Ecology: Washington, DC, USA, 2006.
- Huang, H.; Dane, F.; Kubisiak, T.L. Allozyme and RAPD analysis of the genetic diversity and geographic variation in wild populations of the American chestnut (Fagaceae). Am. J. Bot. 1998, 85, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B. Quaternary history of deciduous forests of eastern North America and Europe. Ann. Mo. Bot. Gard. 1983, 70, 550–563. [Google Scholar] [CrossRef]
- Li, X.; Dane, F. Comparative chloroplast and nuclear DNA analysis of Castanea species in the southern region of the USA. Tree Genet. Genomes 2012, 9, 107–116. [Google Scholar] [CrossRef]
- Shorthouse, D. SimpleMappr, an Online Tool to Produce Publication-Quality Point Maps. Available online: http://www.simplemappr.net (accessed on 27 April 2018).
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, M.J.; Carpten, J.D.; Smith, J.R. Modulation of non-templated nucleotide addition by Taq DNA polymerase: Primer modifications that facilitate genotyping. BioTechniques 1996, 20, 1004–1006, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 2 July 2018).
- Revelle, W. Psych: Procedures for Personality and Psychological Research. Available online: https://CRAN.R-project.org/package=psych (accessed on 31 October 2018).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform. 2008, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Rellstab, C.; Gugerli, F.; Eckert, A.J.; Hancock, A.M.; Holderegger, R. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 2015, 24, 4348–4370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narum, S.R.; Hess, J.E. Comparison of FST outlier tests for SNP loci under selection. Mol. Ecol. Resour. 2011, 11, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [PubMed]
- Ge, Y.; Dudoit, S.; Speed, T.P. Resampling-based multiple testing for microarray data analysis. Test 2003, 12, 1–77. [Google Scholar] [CrossRef] [Green Version]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. B Biol. Sci. 1996, 263, 1619–1626. [Google Scholar] [CrossRef]
- Zhan, X.; Dixon, A.; Batbayar, N.; Bragin, E.; Ayas, Z.; Deutschova, L.; Chavko, J.; Domashevsky, S.; Dorosencu, A.; Bagyura, J.; et al. Exonic versus intronic SNPs: Contrasting roles in revealing the population genetic differentiation of a widespread bird species. Heredity 2015, 114, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Henry, P.; Russello, M.A. Adaptive divergence along environmental gradients in a climate-change-sensitive mammal. Ecol. Evol. 2013, 3, 3906–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Whitlock, R.; Press, M.C.; Scholes, J.D. Variation for host range within and among populations of the parasitic plant Striga hermonthica. Heredity 2012, 108, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Lind, B.M.; Menon, M.; Bolte, C.E.; Faske, T.M.; Eckert, A.J. The genomics of local adaptation in trees: Are we out of the woods yet? Tree Genet. Genomes 2018, 14. [Google Scholar] [CrossRef]
- Lander, E.; Schork, N. Genetic dissection of complex traits. Science 1994, 265, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Marinoni, D.; Akkak, A.; Bounous, G.; Edwards, K.J.; Botta, R. Development and characterization of microsatellite markers in Castanea sativa (Mill.). Mol. Breed. 2003, 11, 127–136. [Google Scholar] [CrossRef]
- McEwan, R.W.; Keiffer, C.H.; McCarthy, B.C. Dendroecology of American chestnut in a disjunct stand of oak–chestnut forest. Can. J. For. Res. 2006, 36, 1–11. [Google Scholar] [CrossRef]
- Schaberg, P.G.; Saielli, T.M.; Hawley, G.J.; Halman, J.M.; Gurney, K.M. Winter Injury of American Chestnut Seedlings Grown in A Common Garden at The Species’ Northern Range Limit; Miller, G.W., Schuler, T.M., Gottschalk, K.W., Brooks, J.R., Grushecky, S.T., Spong, B.D., Rentch, J.S., Eds.; U.S. Department of Agriculture, Forest Service, Northern Research Station: Morgantown, WV, USA, 2013; pp. 72–79.
- Saielli, T.M.; Schaberg, P.G.; Hawley, G.J.; Halman, J.M.; Gurney, K.M. Nut cold hardiness as a factor influencing the restoration of American chestnut in northern latitudes and high elevations. Can. J. For. Res. 2012, 42, 849–857. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Population | Number of Individuals | Latitude | Longitude | Altitude [m a.s.l.] | Annual Mean Temperature [°C] a | Annual Precipitation [mm] a | Mean Growing Season Temperature [°C] a | Mean Growing Season Precipitation [mm] a |
|---|---|---|---|---|---|---|---|---|
| Ontario | 32 | 43.08 | 80.3 | 231 | 7.3 | 921 | 16.98 | 404 |
| Massachusetts | 32 | 42.22 | 72.31 | 111 | 8.3 | 1119 | 17.78 | 484 |
| New York | 26 | 41.44 | 74.13 | 110 | 8.2 | 1207 | 17.66 | 533 |
| Portland | 25 | 41.35 | 72.37 | 2 | 9.6 | 1222 | 18.70 | 504 |
| Pennsylvania | 31 | 39.48 | 76.59 | 119 | 11.2 | 1032 | 20.38 | 464 |
| Maryland | 31 | 39.37 | 79.07 | 569 | 7.9 | 1120 | 16.5 | 513 |
| Kentucky | 32 | 37.50 | 83.51 | 213 | 12.4 | 1214 | 20.98 | 547 |
| Asheville | 31 | 35.46 | 82.10 | 300 | 10.7 | 1435 | 17.94 | 628 |
| Murphy | 32 | 35.05 | 84.01 | 540 | 13.3 | 1491 | 20.94 | 598 |
| Variable | Description | PC1 | PC2 | PC3 |
|---|---|---|---|---|
| Correlation Coefficient | ||||
| Longitude | longitude | 0.62 | 0.23 | −0.65 |
| Latitude | latitude | −0.93 | 0.10 | 0.23 |
| GST | mean growing season temperature | 0.73 | 0.57 | 0.33 |
| GSP | mean growing season precipitation | 0.78 | −0.38 | −0.18 |
| Altitude | altitude | 0.30 | −0.13 | −0.80 |
| bio1 | annual mean temperature | 0.85 | 0.38 | 0.27 |
| bio2 | mean diurnal range | 0.76 | 0.13 | −0.05 |
| bio3 | isothermality | 0.85 | −0.05 | −0.22 |
| bio4 | temperature seasonality | −0.93 | 0.25 | 0.22 |
| bio5 | max. temperature of warmest month | 0.35 | 0.84 | 0.40 |
| bio6 | min. temperature of coldest month | 0.97 | 0.07 | −0.10 |
| bio7 | temperature annual range | −0.79 | 0.51 | 0.31 |
| bio8 | mean temperature of wettest quarter | −0.48 | 0.45 | −0.59 |
| bio9 | mean temperature of driest quarter | 0.92 | 0.23 | 0.18 |
| bio10 | mean temperature of warmest quarter | 0.55 | 0.67 | 0.43 |
| bio11 | mean temperature of coldest quarter | 0.98 | 0.03 | 0.00 |
| bio12 | annual precipitation | 0.87 | −0.33 | 0.12 |
| bio13 | precipitation of wettest month | 0.83 | −0.27 | −0.03 |
| bio14 | precipitation of driest month | 0.62 | −0.65 | 0.32 |
| bio15 | precipitation seasonality | 0.34 | 0.37 | −0.77 |
| bio16 | precipitation of wettest quarter | 0.80 | −0.30 | −0.17 |
| bio17 | precipitation of driest quarter | 0.75 | −0.47 | 0.27 |
| bio18 | precipitation of warmest quarter | 0.80 | −0.40 | −0.27 |
| bio19 | precipitation of coldest quarter | 0.83 | −0.28 | 0.23 |
| Complete Marker Set | g-SSRs | EST-SSRs | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Population | Na | Pa | Ho | He | FIS | Na | Pa | Ho | He | FIS | Na | Pa | Ho | He | FIS |
| Ontario | 6.2 | 0.375 | 0.550 | 0.578 | 0.0661 * | 8.4 | 0.000 | 0.729 | 0.727 | 0.0166 * | 5.3 | 0.529 | 0.477 | 0.517 | 0.0930 * |
| Massachusetts | 5.0 | 0.042 | 0.560 | 0.575 | 0.0406 * | 7.9 | 0.143 | 0.706 | 0.783 | 0.1117 * | 3.9 | 0.000 | 0.499 | 0.489 | −0.0050 |
| New York | 5.1 | 0.042 | 0.585 | 0.556 | −0.0328 | 7.6 | 0.143 | 0.753 | 0.704 | −0.0493 | 4.1 | 0.000 | 0.516 | 0.495 | −0.0230 |
| Portland | 4.1 | 0.000 | 0.526 | 0.469 | −0.1011 | 5.4 | 0.000 | 0.743 | 0.651 | −0.1207 | 3.5 | 0.000 | 0.437 | 0.394 | −0.0877 |
| Pennsylvania | 5.1 | 0.042 | 0.562 | 0.542 | −0.0227 * | 6.6 | 0.000 | 0.736 | 0.706 | −0.0349 * | 4.5 | 0.059 | 0.490 | 0.475 | −0.0154 |
| Maryland | 5.8 | 0.042 | 0.563 | 0.553 | −0.0021 | 9.1 | 0.143 | 0.738 | 0.743 | 0.0220 | 4.5 | 0.000 | 0.491 | 0.475 | −0.0174 |
| Kentucky | 5.8 | 0.208 | 0.521 | 0.565 | 0.0945 * | 8.4 | 0.143 | 0.618 | 0.728 | 0.1668 * | 4.7 | 0.235 | 0.481 | 0.498 | 0.0508 * |
| Asheville | 5.9 | 0.125 | 0.530 | 0.545 | 0.0433 * | 8.9 | 0.143 | 0.720 | 0.731 | 0.0265 | 4.7 | 0.118 | 0.451 | 0.468 | 0.0525 * |
| Murphy | 7.3 | 0.833 | 0.586 | 0.610 | 0.0552 * | 10.7 | 0.857 | 0.753 | 0.801 | 0.0782 * | 5.8 | 0.824 | 0.517 | 0.531 | 0.0413 * |
| Mean | 5.6 | 0.190 | 0.554 | 0.555 | 0.0157 | 8.1 | 0.175 | 0.722 | 0.730 | 0.0241 | 4.5 | 0.196 | 0.484 | 0.482 | 0.0099 |
| Marker | Na | Ho | He | FIS |
|---|---|---|---|---|
| CmSI0031 | 7.1 | 0.696 | 0.719 | 0.0534 * |
| CmSI0049 | 3.4 | 0.243 | 0.231 | −0.0331 * |
| CmSI0327 | 7.0 | 0.731 | 0.743 | 0.0333 |
| CmSI0391 | 4.0 | 0.585 | 0.585 | 0.0190 |
| CmSI0396 | 3.7 | 0.596 | 0.601 | 0.0305 |
| CmSI0495 | 3.9 | 0.350 | 0.350 | 0.0258 |
| CmSI0527 | 3.6 | 0.272 | 0.292 | 0.0851 |
| CmSI0537 | 4.2 | 0.235 | 0.249 | 0.0721 |
| CmSI0551 | 4.2 | 0.344 | 0.368 | 0.0883 |
| CmSI0559 | 3.8 | 0.616 | 0.601 | −0.0079 |
| CmSI0561 | 5.0 | 0.477 | 0.468 | 0.0056 |
| CmSI0594 | 3.3 | 0.365 | 0.375 | 0.0524 |
| CmSI0600 | 8.7 | 0.809 | 0.771 | −0.0331 |
| CmSI0608 | 2.0 | 0.415 | 0.372 | −0.1021 |
| CmSI0611 | 2.4 | 0.113 | 0.118 | 0.0704 |
| CmSI0678 | 5.2 | 0.737 | 0.688 | −0.0528 * |
| CmSI0683 | 5.8 | 0.652 | 0.671 | 0.0474 * |
| CsCAT1 | 8.2 | 0.791 | 0.776 | 0.0048 |
| CsCAT3 | 9.9 | 0.734 | 0.789 | 0.0923 * |
| CsCAT7 | 6.0 | 0.610 | 0.672 | 0.1143 * |
| CsCAT8 | 7.4 | 0.628 | 0.603 | −0.0216 |
| CsCAT14 | 6.8 | 0.767 | 0.712 | −0.0498 * |
| CsCAT24 | 11.7 | 0.785 | 0.855 | 0.0978 * |
| QaCA022 | 6.8 | 0.740 | 0.706 | −0.0289 |
| Marker | PC1 | PC2 | PC3 |
|---|---|---|---|
| CmSI0031 | x | x | |
| CmSI0049 | x | ||
| CmSI0327 | x | x | |
| CmSI0391 | x | ||
| CmSI0594 | x | ||
| CmSI0600 | x | x | x |
| CsCAT1 | x | x | |
| CsCAT3 | x | x | x |
| CsCAT14 | x | ||
| QaCA022 | x |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, M.; Nelson, C.D.; Gailing, O. Analysis of Environment-Marker Associations in American Chestnut. Forests 2018, 9, 695. https://doi.org/10.3390/f9110695
Müller M, Nelson CD, Gailing O. Analysis of Environment-Marker Associations in American Chestnut. Forests. 2018; 9(11):695. https://doi.org/10.3390/f9110695
Chicago/Turabian StyleMüller, Markus, C. Dana Nelson, and Oliver Gailing. 2018. "Analysis of Environment-Marker Associations in American Chestnut" Forests 9, no. 11: 695. https://doi.org/10.3390/f9110695
APA StyleMüller, M., Nelson, C. D., & Gailing, O. (2018). Analysis of Environment-Marker Associations in American Chestnut. Forests, 9(11), 695. https://doi.org/10.3390/f9110695