Evolutionary Constraints to Viroid Evolution


{kind=link}
{kind=link}
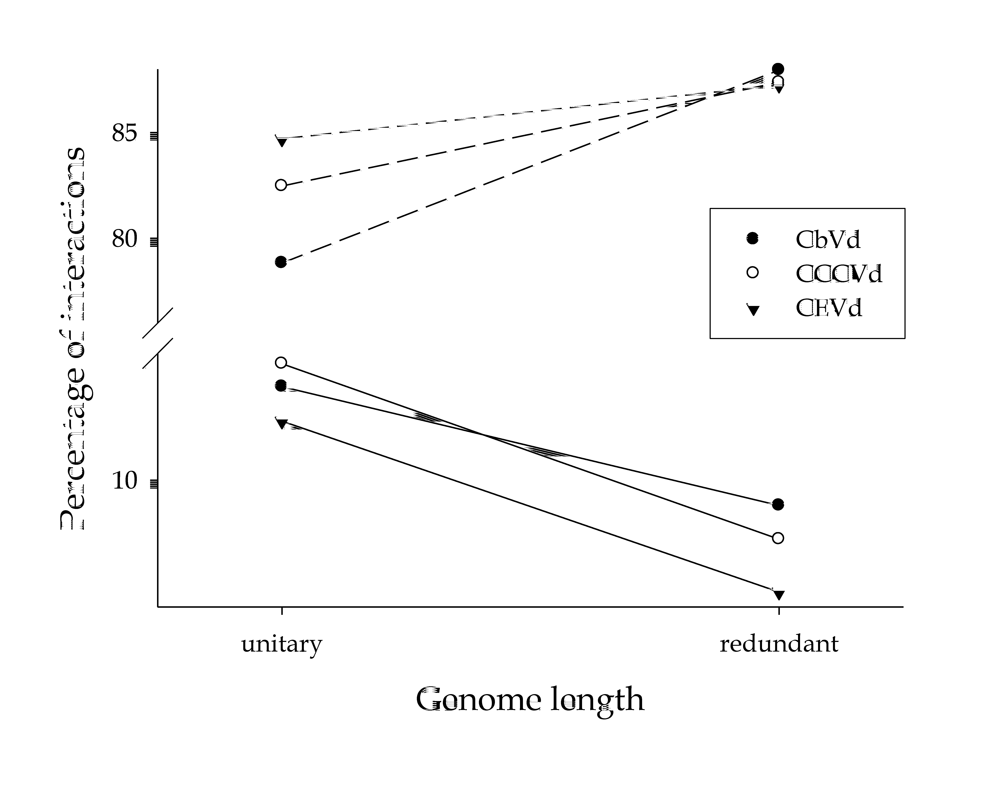{kind=link}
{kind=link}
Abstract
:1. Introduction
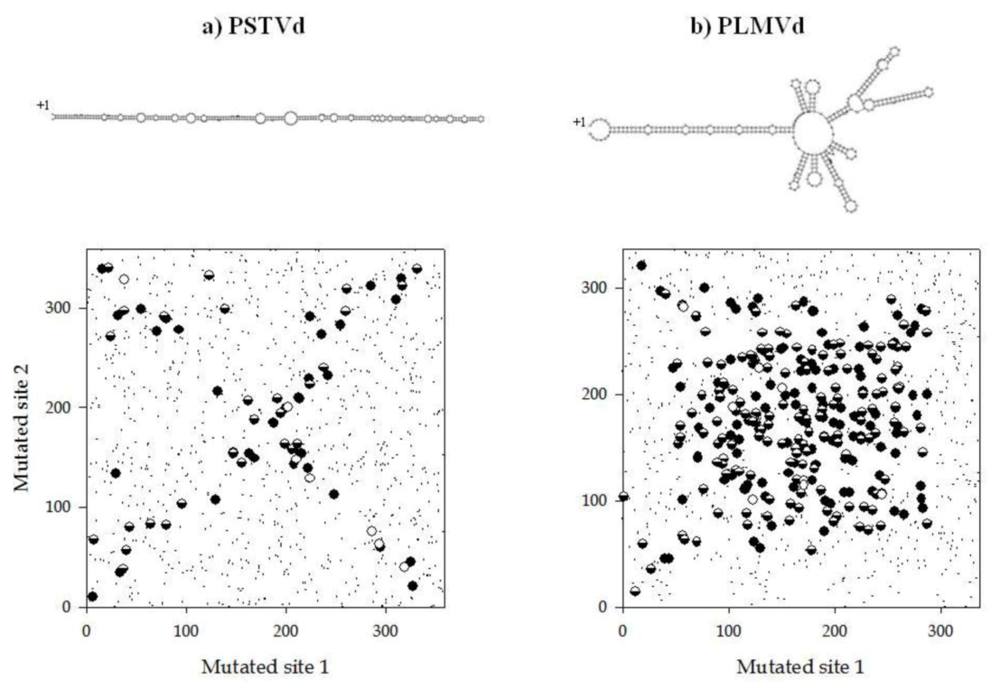
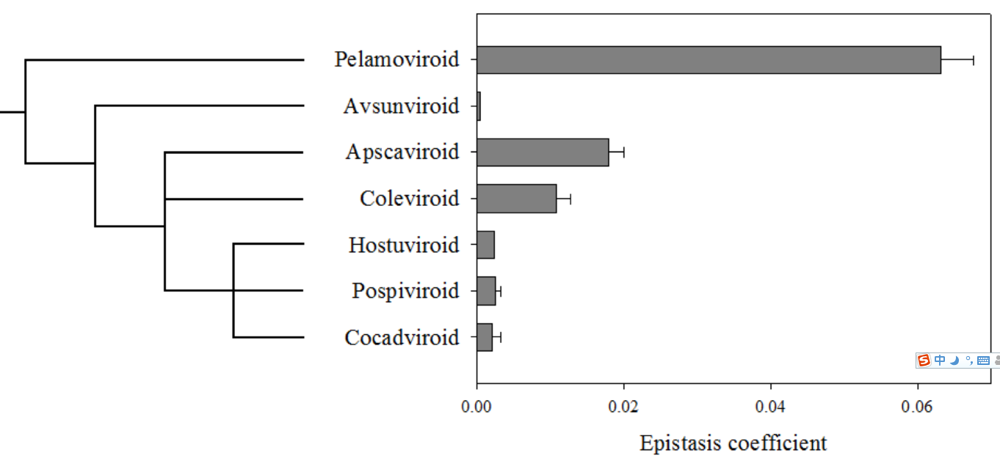
2. Viroids fold into compacted structures as a way to avoid RNA silencing

3. A compact RNA folding means epistasis

3.1. Two ways to escape from the tyranny of antagonistic epistasis: genetic redundancy and recombination


4. Small genomes and high mutation rates: the Eigen’s paradox
5. Conclusions
Acknowledgments
References and Notes
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Vaucheret, H. Post-transcriptional small RNA pathways in plants: mechanisms and regulations. Genes Dev. 2006, 20, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ferrer, V.; Voinnet, O. Roles of plant small RNAs in biotic stress responses. Annu. Rev. Plant Biol. 2009, 60, 485–510. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Morris, T.J. Suppressors of RNA silencing encoded by plant viruses and their role in viral infections. FEBS Lett. 2005, 579, 5958–5964. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Provost, P.; Taylor, J.M. Resistance of human hepatitis delta virus RNAs to Dicer activity. J. Virol. 2003, 77, 11910–11917. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Hernández, C.; Martínez de Alba, A.E.; Daròs, J.A.; di Serio, F. Viroids and viroid-host interactions. Annu. Rev. Phytopathol. 2005, 43, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Vogt, U.; Pelissier, T.; Putz, A.; Razvi, F.; Fischer, R.; Wassenegger, M. Viroid-induced RNA silencing of GFP-viroid fusion transgenes does not induce extensive spreading of methylation or transitive silencing. Plant J. 2004, 38, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.B.; Bian, X.Y.; Wu, L.M.; Liu, L.X.; Smith, N.A.; Isenegger, D.; Wu, R.M.; Masuta, C.; Vance, V.B.; Watson, J.M.; Rezaian, A.; Dennis, E.S.; Waterhouse, P.M. On the role of RNA silencing in the pathogenicity and evolution of viroids and viral satellites. Proc. Natl. Acad. Sci. USA 2004, 101, 3275–3280. [Google Scholar] [CrossRef]
- Itaya, A.; Zhong, X.; Bundschuh, R.; Qi, Y.; Wang, Y.; Takeda, R.; Harris, A.R.; Molina, C.; Nelson, R.S.; Ding, B. A structured viroid RNA Is substrate for Dicer-like cleavage to produce biologically active small RNAs but is resistant to RISC-mediated degradation. J. Virol. 2007, 81, 2980–2994. [Google Scholar] [CrossRef] [PubMed]
- Gómez, G.; Pallás, V. Mature monomeric forms of Hop stunt viroid resist RNA silencing in transgenic plants. Plant J. 2007, 51, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Stone, J.K.; Andino, R. Poliovirus escape from RNA interference: short interfering RNA-target recognition and implications for therapeutic approaches. J. Virol. 2005, 79, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer-Kazemi, F.R.; Sczakiel, G. The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucl. Acids Res. 2003, 31, 4417–4424. [Google Scholar] [CrossRef]
- Yoshinari, K.; Miyagishi, M.; Taira, K. Effects on RNAi of the tight structure, sequence and position of the targeted region. Nucl. Acids Res. 2004, 32, 691–696. [Google Scholar] [CrossRef]
- Westerhout, E.M.; Ooms, M.; Vink, M.; Das, A.T.; Berkhout, B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucl. Acids Res. 2005, 33, 796–804. [Google Scholar] [CrossRef]
- Westerhout, E.M.; Berkhout, B. A systematic analysis of the effect of target RNA structure on RNA interference. Nucl. Acids Res. 2007, 35, 4322–4330. [Google Scholar] [CrossRef]
- Fadda, Z.; Daròs, J.A.; Flores, R.; Durán-Vila, N. Identification in eggplant of a variant of Citrus exocortis viroid (CEVd) with a 96 nucleotide duplication in the right terminal region of the rod-like secondary structure. Virus Res. 2003, 97, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, M.; di Serio, F.; Covelli, L.; Ragozzino, A.; Hernández, C.; Flores, R. Peach latent mosaic viroid variants inducing peach calico (extreme chlorosis) contain a characteristic insertion that is responsible for this symptomatology. Virology 2003, 313, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, D.M.; Watson, R.A.; Chao, L. Sign epistasis and genetic constraint on evolutionary trajectories. Evolution 2005, 59, 1165–1174. [Google Scholar] [PubMed]
- Sanjuán, R.; Elena, S.F. Epistasis correlates to genomic complexity. Proc. Natl. Acad. Sci. USA 2006, 103, 14402–14405. [Google Scholar] [CrossRef]
- Sanjuán, R.; Forment, J.; Elena, S.F. In silico predicted robustness of viroid RNA secondary structures. II. Interaction between mutation pairs. Mol. Biol. Evol. 2006, 23, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Cuevas, J.M.; Moya, A.; Elena, S.F. Epistasis and adaptability of an RNA virus. Genetics 2005, 170, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Bernard, L.; Duran-Vila, N.; Elena, S.F. Effect of citrus hosts on the generation, maintenance and evolutionary fate of genetic variability of Citrus exocortis viroid. J. Gen. Virol. 2009, 90, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Gros, P.A.; Le Nagard, H.; Tenaillon, O. The evolution of epistasis and its links with genetic robustness, complexity and drift in a phenotypic model of adaptation. Genetics 2009, 182, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Nebot, M.R. A network model for the correlation between epistasis and genomic complexity. PLoS ONE 2008, 3, e2663. [Google Scholar] [CrossRef] [PubMed]
- Proulx, S.R.; Phillips, P.C. The opportunity for canalization and the evolution of genetic networks. Am. Nat. 2005, 165, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Tyler, A.L.; Asselbergs, F.W.; Williams, S.M.; Moore, J.H. Shadows of complexity: what biological networks reveal about epistasis and pleiotropy. BioEssays 2009, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Visvander, J.E.; Symons, R.H. Comparative sequence and structure of different isolates of Citrus exocortis viroid. Virology 1983, 130, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Haseloff, J.; Mohammed, N.A.; Symons, R.H. Viroids RNAs of cadang-cadang disease of coconuts. Nature 1982, 299, 316–321. [Google Scholar] [CrossRef]
- Singh, R.P.; Boiteux, M.E.N.F.; Ready, K.F.M.; Nie, X. Coleus blumei viroid. In Viroids; Hadidi, A., Flores, F., Randles, J.W., Semancik, J.S., Eds.; CSIRO: Collingwood, Australia, 2003. [Google Scholar]
- de Visser, J.A.G.M.; Elena, S.F. The evolution of sex: empirical insights into the roles of epistasis and drift. Nat. Rev. Genet. 2007, 8, 139–149. [Google Scholar] [CrossRef]
- Misevic, D.; Ofria, C.; Lenski, R.E. Sexual reproduction reshapes the genetic architecture of digital organisms. Proc. R. Soc. B 2005, 273, 457–464. [Google Scholar] [CrossRef]
- Keese, P.; Symons, R.H. Domains in viroids: evidence of intermolecular RNA rearrangements and their contribution to viroid evolution. Proc. Natl. Acad. Sci. USA 1985, 82, 4582–4586. [Google Scholar] [CrossRef]
- Hammond, R.; Smith, D.R.; Diener, T.O. Nucleotide sequence and proposed secondary structure of Columnea latent viroid a natural mosaic of viroid sequences. Nucl. Acids Res. 1989, 17, 10083–10094. [Google Scholar] [CrossRef]
- Puchta, H.; Ramm, K.; Luckinger, R.; Hadas, R.; Bar-Joseph, M.; Sanger, H.L. Primary and secondary structure of Citrus viroid IV (CVd IV), a new chimeric viroid present in dwarfed grapefruit in Israel. Nucl. Acids Res. 1991, 19, 6640. [Google Scholar] [CrossRef]
- Rezaian, M.A. Australian grapevine viroid - evidence for extensive recombination between viroids. Nucl. Acids Res. 1990, 18, 1813–1818. [Google Scholar] [CrossRef]
- Elena, S.F.; Dopazo, J.; de la Peña, M.; Flores, R.; Diener, T.O.; Moya, A. Phylogenetic analysis of viroid and viroid-like satellite RNAs from plants: a reassessment. J. Mol. Evol. 2001, 53, 155–159. [Google Scholar] [PubMed]
- Diener, T.O. Subviral pathogens of plants: viroids and viroid-like satellite RNAs. FASEB J. 1991, 5, 2808–2813. [Google Scholar] [PubMed]
- Azevedo, R.B.; Lohaus, R.; Srinivasan, S.; Dang, K.K.; Burch, C.L. Sexual reproduction selects for robustness and negative epistasis in artificial gene networks. Nature 2006, 440, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Carrasco, P.; Daròs, J.A.; Sanjuán, R. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 2006, 7, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Codoñer, F.M.; Daròs, J.A.; Solé, R.V.; Elena, S.F. The fittest versus the flattest: experimental confirmation of the quasispecies effect with subviral pathogens. PLoS Pathog. 2006, 2, e136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakauer, D.C.; Plotkin, J.B. Redundancy, antiredundancy, and the robustness of genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 1405–1409. [Google Scholar] [CrossRef]
- Sanjuán, R.; Forment, J.; Elena, S.F. In silico predicted robustness of viroids RNA secondary structure. I. The effect of single mutations. Mol. Biol. Evol. 2006, 23, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Durán-Vila, N..; Elena, S.F.; Daròs, J.A.; Flores, R. Structure evolution of viroids. In Origin and evolution of viruses, 2nd; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press: London, UK, 2008; pp. 43–64. [Google Scholar]
- Mühlbach, H.P.; Sänger, H.L. Viroid replication is inhibited by α-amanitin. Nature 1979, 278, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Vera, A.; Flores, R. A chloroplastic RNA polymerase resistant to tagetitoxin is involved in replication of Avocado sunblotch viroid. Virology 2000, 268, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuán, R. Extremely high mutation rate of a hammerhead viroid. Science 2009, 323, 1308. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Selforganization of matter and the evolution of macromolecules. Naturwissenschafen 1971, 58, 465–523. [Google Scholar] [CrossRef]
- Elena, S.F.; Sanjuán, R. Adaptive value of high mutation rates of RNA viruses: separating causes from consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef] [PubMed]
- Furió, V.; Moya, A.; Sanjuán, R. The cost of replication fidelity in human immunodeficiency virus type 1. Proc. R. Soc. B 2007, 274, 225–230. [Google Scholar] [CrossRef]
- Furió, V.; Moya, A.; Sanjuán, R. The cost of replication fidelity in an RNA virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10233–10237. [Google Scholar] [CrossRef]
- Clune, J.; Misevic, D.; Ofria, C.; Lenski, R.E.; Elena, S.F.; Sanjuán, R. Natural selection fails to optimize mutation rates for long-term on rugged fitness landscapes. PLoS Comp. Biol. 2008, 4, e1000187. [Google Scholar] [CrossRef] [Green Version]
- Daròs, J.A.; Marcos, J.F.; Hernández, C.; Flores, R. Replication of Avocado sunblotch viroid: evidence for a symmetric pathway with two rolling circles and hammerhead ribozyme processing. Proc. Natl. Acad. Sci. USA 1994, 91, 12813–12817. [Google Scholar] [CrossRef]
- Zhong, X.; Archual, A.J.; Amin, A.A.; Ding, B. A genomic map of viroid RNA motifs critical for replication and systemic trafficking. Plant Cell 2008, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Elena, S.F.; Gómez, G.; Daròs, J.-A. Evolutionary Constraints to Viroid Evolution. Viruses 2009, 1, 241-254. https://doi.org/10.3390/v1020241
Elena SF, Gómez G, Daròs J-A. Evolutionary Constraints to Viroid Evolution. Viruses. 2009; 1(2):241-254. https://doi.org/10.3390/v1020241
Chicago/Turabian StyleElena, Santiago F., Gustavo Gómez, and José-Antonio Daròs. 2009. "Evolutionary Constraints to Viroid Evolution" Viruses 1, no. 2: 241-254. https://doi.org/10.3390/v1020241
APA StyleElena, S. F., Gómez, G., & Daròs, J. -A. (2009). Evolutionary Constraints to Viroid Evolution. Viruses, 1(2), 241-254. https://doi.org/10.3390/v1020241