HCV Innate Immune Responses

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Interferons
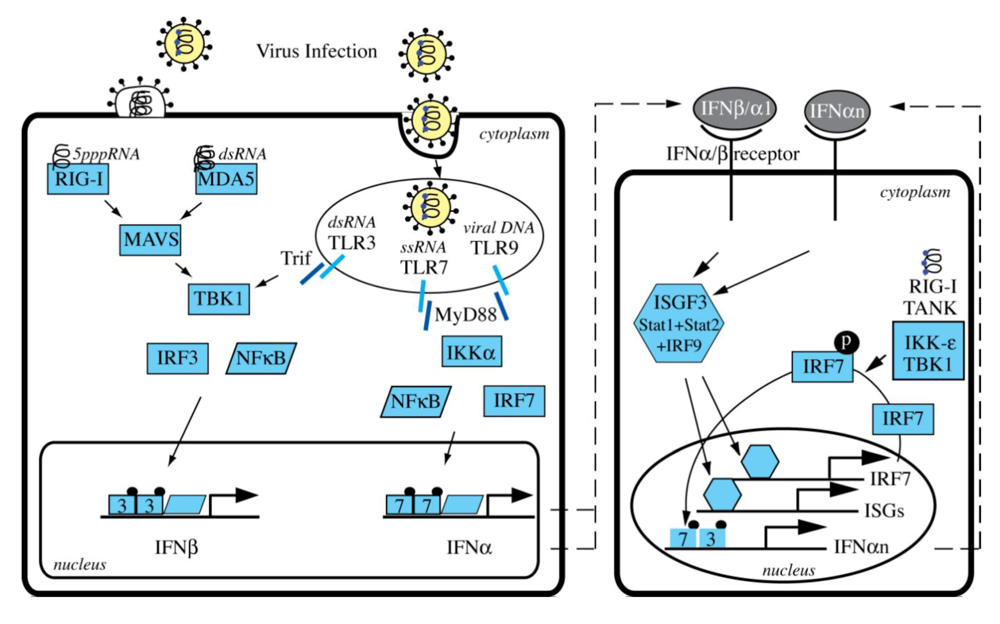
3. Induction of Type I Interferons

4. HCV Interference with Cellular Sensors
5. Induction of Interferon Stimulated Genes by HCV
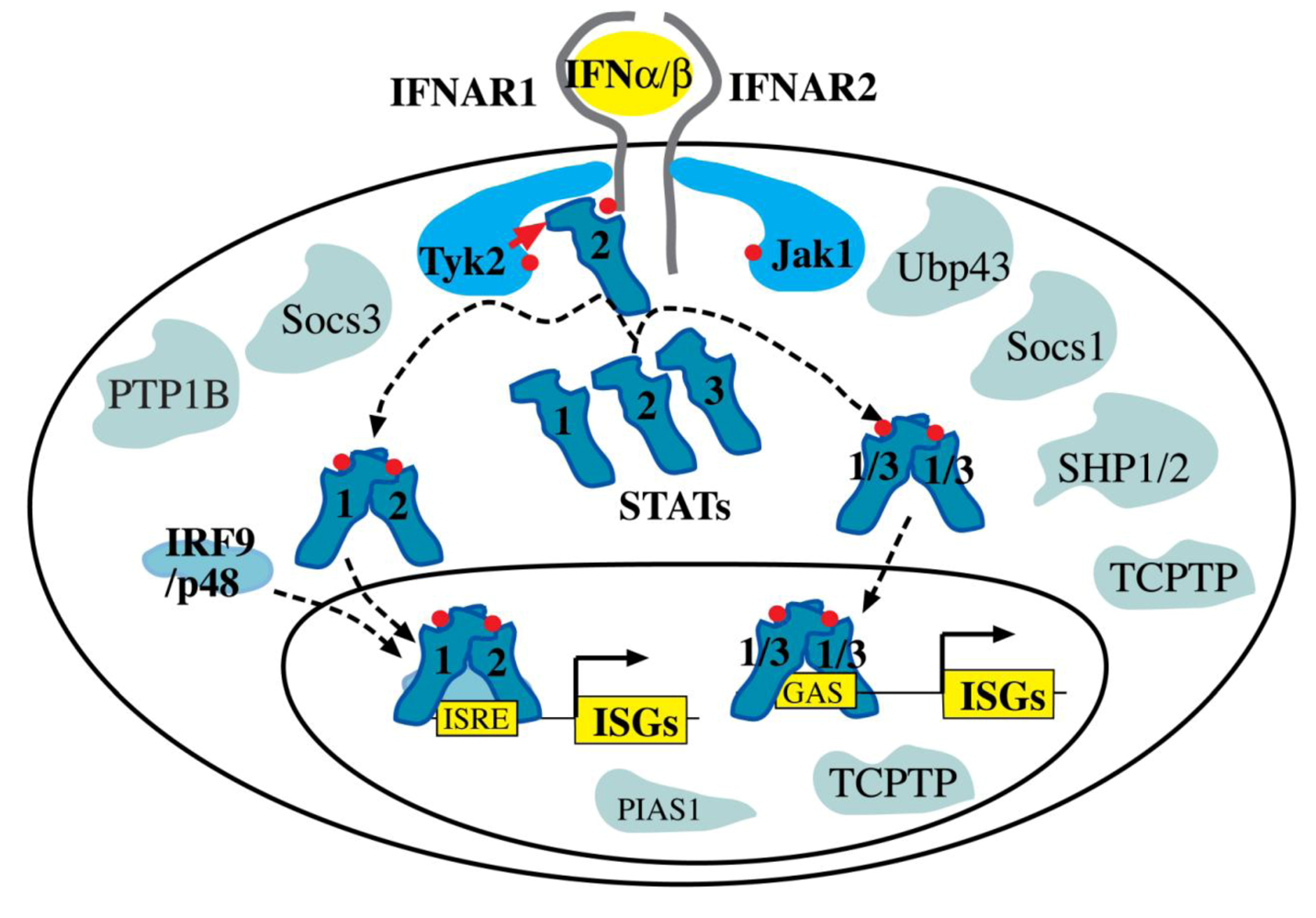
6. Interferon signaling through the Jak-STAT pathway
6.1. The receptor-kinase complex
6.2. Signal Transducers and Activators of Transcription (STATs)

6.3. Negative Regulators of Interferon Signaling
6.3.1. Suppressor of Cytokine Signaling (SOCS)
6.3.2. USP18
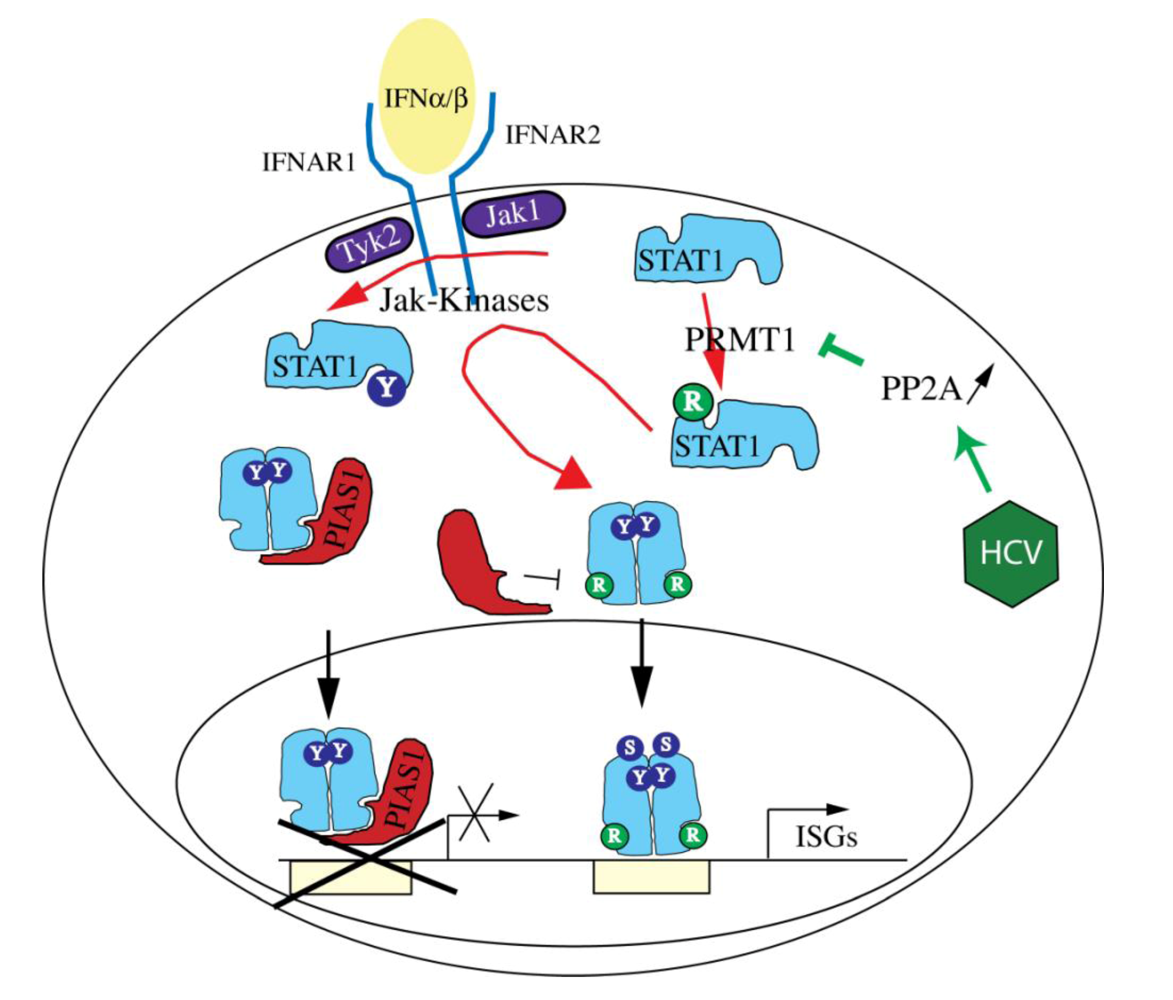
6.3.3. Protein Inhibitor of Activated STAT1 (PIAS1) and PIAS3
6.3.4. TcPTP
7. Interference of HCV with IFN Signaling through the Jak-STAT pathway

8. Effects of Type I Interferons
8.1. Interferon Regulated Genes
9. Interference of Hepatitis C Virus with Interferon Effector Systems
10. Conclusions
References
- Isaacs, A.; Lindenmann, J. Virus interferenceI. The interferon. Proc. R. Soc. Lond. B. Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Uze, G.; Lutfalla, G.; Gresser, I. Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell 1990, 60, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Novick, D.; Cohen, B.; Rubinstein, M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell 1994, 77, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Lutfalla, G.; Holland, S.J.; Cinato, E.; Monneron, D.; Reboul, J.; Rogers, N.C.; Smith, J.M.; Stark, G.R.; Gardiner, K.; Mogensen, K.E.; et al Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. Embo J. 1995, 14, 5100–5108. [Google Scholar] [PubMed]
- Aguet, M.; Dembic, Z.; Merlin, G. Molecular cloning and expression of the human interferon-gamma receptor. Cell 1988, 55, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.; Donnelly, R.J.; Kotenko, S.; Mariano, T.M.; Cook, J.R.; Wang, N.; Emanuel, S.; Schwartz, B.; Miki, T.; Pestka, S. Identification and sequence of an accessory factor required for activation of the human interferon gamma receptor. Cell 1994, 76, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, S.; Bohni, R.; Stark, G.; Di Marco, F.; Aguet, M. A novel member of the interferon receptor family complements functionality of the murine interferon gamma receptor in human cells. Cell 1994, 76, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Severa, M.; Giacomini, E.; Monneron, D.; Remoli, M.E.; Julkunen, I.; Cella, M.; Lande, R.; Uze, G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004, 34, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Sheikh, F.; Kotenko, S.V.; Dickensheets, H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J. Leukoc. Biol. 2004, 76, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; Sulkowski, M.; McHutchison, J.G.; Goldstein, D.B. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nishida, N.; Sugiyama, M.; Kurosaki, M.; Matsuura, K.; Sakamoto, N.; Nakagawa, M.; Korenaga, M.; Hino, K.; Hige, S.; Ito, Y.; Mita, E.; Tanaka, E.; Mochida, S.; Murawaki, Y.; Honda, M.; Sakai, A.; Hiasa, Y.; Nishiguchi, S.; Koike, A.; Sakaida, I.; Imamura, M.; Ito, K.; Yano, K.; Masaki, N.; Sugauchi, F.; Izumi, N.; Tokunaga, K.; Mizokami, M. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009, 41, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; Riordan, S.; Sheridan, D.; Smedile, A.; Fragomeli, V.; Muller, T.; Bahlo, M.; Stewart, G.J.; Booth, D.R.; George, J. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. Function of RIG-I-like receptors in antiviral innate immunity. J. Biol. Chem. 2007, 282, 15315–15318. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Kirschning, C.J.; Hacker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. Embo J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, A.; Barnes, B.J.; Amrute, S.; Yeow, W.S.; Megjugorac, N.; Dai, J.; Feng, D.; Chung, E.; Pitha, P.M.; Fitzgerald-Bocarsly, P. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J. Leukoc. Biol. 2003, 74, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Kerkmann, M.; Rothenfusser, S.; Hornung, V.; Towarowski, A.; Wagner, M.; Sarris, A.; Giese, T.; Endres, S.; Hartmann, G. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J. Immunol. 2003, 170, 4465–4474. [Google Scholar] [PubMed]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; Takeuchi, O.; Akira, S. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef]
- Hoshino, K.; Sugiyama, T.; Matsumoto, M.; Tanaka, T.; Saito, M.; Hemmi, H.; Ohara, O.; Akira, S.; Kaisho, T. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 2006, 440, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell. 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K. J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale Jr., M.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef]
- Sumpter Jr., R.; Loo, Y.M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale Jr., M. J. Virol. 2005, 79, 2689–2699. [CrossRef] [PubMed]
- Bigger, C.B.; Brasky, K.M.; Lanford, R.E. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 2001, 75, 7059–7066. [Google Scholar] [CrossRef] [PubMed]
- Bigger, C.B.; Guerra, B.; Brasky, K.M.; Hubbard, G.; Beard, M.R.; Luxon, B.A.; Lemon, S.M.; Lanford, R.E. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 2004, 78, 13779–13792. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Borozan, I.; Feld, J.; Sun, J.; Tannis, L.L.; Coltescu, C.; Heathcote, J.; Edwards, A.M.; McGilvray, I.D. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology 2005, 128, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Sarasin-Filipowicz, M.; Oakeley, E.J.; Duong, F.H.; Christen, V.; Terracciano, L.; Filipowicz, W.; Heim, M.H. Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl. Acad. Sci. USA 2008, 105, 7034–7039. [Google Scholar] [CrossRef]
- Asselah, T.; Bieche, I.; Narguet, S.; Sabbagh, A.; Laurendeau, I.; Ripault, M.P.; Boyer, N.; Martinot-Peignoux, M.; Valla, D.; Vidaud, M.; Marcellin, P. Liver gene expression signature to predict response to pegylated interferon plus ribavirin combination therapy in patients with chronic hepatitis C. Gut 2008, 57, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Darnell Jr., J.E. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Darnell Jr., J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [PubMed]
- Heim, M.H. The STAT protein family. Sehgal P. B.; Levy D. E.;, Hirano, T., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands/Boston, USA/London, UK, 2003; pp. 11–26. [Google Scholar]
- Krebs, D.L.; Hilton, D.J. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Song, M.M.; Shuai, K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J. Biol. Chem. 1998, 273, 35056–35062. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Kinjyo, I.; Yoshimura, A. The janus kinase inhibitor, Jab/SOCS-1, is an interferon-gamma inducible gene and determines the sensitivity to interferons. Leuk. Lymphoma 2000, 38, 49–58. [Google Scholar] [PubMed]
- Alexander, W.S.; Starr, R.; Fenner, J.E.; Scott, C.L.; Handman, E.; Sprigg, N.S.; Corbin, J.E.; Cornish, A.L.; Darwiche, R.; Owczarek, C.M.; Kay, T.W.; Nicola, N.A.; Hertzog, P.J.; Metcalf, D.; Hilton, D.J. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 1999, 98, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Fenner, J.E.; Starr, R.; Cornish, A.L.; Zhang, J.G.; Metcalf, D.; Schreiber, R.D.; Sheehan, K.; Hilton, D.J.; Alexander, W.S.; Hertzog, P.J. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat. Immunol. 2006, 7, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Q.; Ilaria Jr., R.; Kingsley, P.D.; Iwama, A.; van Etten, R.A.; Palis, J.; Zhang, D.E. A novel ubiquitin-specific protease, UBP43, cloned from leukemia fusion protein AML1-ETO-expressing mice, functions in hematopoietic cell differentiation. Mol. Cell. Biol. 1999, 19, 3029–3038. [Google Scholar] [PubMed]
- Malakhov, M.P.; Malakhova, O.A.; Kim, K.I.; Ritchie, K.J.; Zhang, D.E. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J. Biol. Chem. 2002, 277, 9976–9981. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. Embo J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.J.; Malakhov, M.P.; Hetherington, C.J.; Zhou, L.; Little, M.T.; Malakhova, O.A.; Sipe, J.C.; Orkin, S.H.; Zhang, D.E. Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev. 2002, 16, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Yan, M.; Malakhov, M.P.; Yuan, Y.; Ritchie, K.J.; Kim, K.I.; Peterson, L.F.; Shuai, K.; Zhang, D.E. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003, 17, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.J.; Hahn, C.S.; Kim, K.I.; Yan, M.; Rosario, D.; Li, L.; de la Torre, J. C.; Zhang, D.E. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 2004, 10, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Sarasin-Filipowicz, M.; Wang, X.; Yan, M.; Duong, F.H.; Poli, V.; Hilton, D.J.; Zhang, D.E.; Heim, M.H. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol. Cell. Biol. 2009, 29, 4841–4851. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liao, J.; Rao, X.; Kushner, S.A.; Chung, C.D.; Chang, D.D.; Shuai, K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. USA 1998, 95, 10626–10631. [Google Scholar] [CrossRef]
- Liu, B.; Mink, S.; Wong, K.A.; Stein, N.; Getman, C.; Dempsey, P.W.; Wu, H.; Shuai, K. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 2004, 5, 891–898. [Google Scholar] [CrossRef]
- ten Hoeve, J.; de Jesus Ibarra-Sanchez, M.; Fu, Y.; Zhu, W.; Tremblay, M.; David, M.; Shuai, K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 2002, 22, 5662–5668. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, K.M.; Nestel, F.P.; Newell, E.W.; Charette, G.; Seemayer, T.A.; Tremblay, M.L.; Lapp, W.S. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood 2004, 103, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Weber, F.; Haller, O. Viral suppression of the interferon system. Biochimie 2007, 89, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Brenndorfer, E.D.; Haussinger, D. Hepatitis C virus (HCV) employs multiple strategies to subvert the host innate antiviral response. Biol. Chem. 2008, 389, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Ludwig, S.; Ehrhardt, C.; Albrecht, U.; Erhardt, A.; Schaper, F.; Heinrich, P.C.; Haussinger, D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. Faseb J. 2003, 17, 488–490. [Google Scholar] [PubMed]
- Kawaguchi, T.; Yoshida, T.; Harada, M.; Hisamoto, T.; Nagao, Y.; Ide, T.; Taniguchi, E.; Kumemura, H.; Hanada, S.; Maeyama, M.; Baba, S.; Koga, H.; Kumashiro, R.; Ueno, T.; Ogata, H.; Yoshimura, A.; Sata, M. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 2004, 165, 1499–1508. [Google Scholar] [PubMed]
- Lin, W.; Choe, W.H.; Hiasa, Y.; Kamegaya, Y.; Blackard, J.T.; Schmidt, E.V.; Chung, R.T. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology 2005, 128, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Melen, K.; Fagerlund, R.; Nyqvist, M.; Keskinen, P.; Julkunen, I. Expression of hepatitis C virus core protein inhibits interferon-induced nuclear import of STATs. J. Med. Virol. 2004, 73, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Larrea, E.; Aldabe, R.; Molano, E.; Fernandez-Rodriguez, C.M.; Ametzazurra, A.; Civeira, M.P.; Prieto, J. Altered expression and activation of signal transducers and activators of transcription (STATs) in hepatitis C virus infection: in vivo and in vitro studies. Gut 2006, 55, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Moradpour, D.; Blum, H.E. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J. Virol. 1999, 73, 8469–8475. [Google Scholar] [PubMed]
- Blindenbacher, A.; Duong, F.H.; Hunziker, L.; Stutvoet, S.T.; Wang, X.; Terracciano, L.; Moradpour, D.; Blum, H.E.; Alonzi, T.; Tripodi, M.; La Monica, N.; Heim, M.H. Expression of hepatitis c virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology 2003, 124, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Duong, F.H.; Filipowicz, M.; Tripodi, M.; La Monica, N.; Heim, M.H. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology 2004, 126, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Christen, V.; Treves, S.; Duong, F.H.; Heim, M.H. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology 2007, 46, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Duong, F.H.; Christen, V.; Berke, J.M.; Penna, S.H.; Moradpour, D.; Heim, M.H. Upregulation of Protein Phosphatase 2Ac by Hepatitis C Virus Modulates NS3 Helicase Activity through Inhibition of Protein Arginine Methyltransferase 1. J. Virol. 2005, 79, 15342–15350. [Google Scholar] [CrossRef] [PubMed]
- Mowen, K.A.; Tang, J.; Zhu, W.; Schurter, B.T.; Shuai, K.; Herschman, H.R.; David, M. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell 2001, 104, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Meissner, T.; Krause, E.; Lodige, I.; Vinkemeier, U. Arginine methylation of STAT1: a reassessment. Cell 2004, 119, 587–590. [Google Scholar] [PubMed]
- Duong, F.H.; Christen, V.; Filipowicz, M.; Heim, M.H. S-adenosylmethionine and betaine correct hepatitis C virus induced inhibition of interferon signaling in vitro. Hepatology 2006, 43, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.C.; Haque, S.J.; Williams, B.R. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. Embo J. 1999, 18, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sassano, A.; Majchrzak, B.; Deb, D.K.; Levy, D.E.; Gaestel, M.; Nebreda, A.R.; Fish, E.N.; Platanias, L.C. Role of p38alpha Map kinase in Type I interferon signaling. J. Biol. Chem. 2004, 279, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Pfeffer, S.R.; Kim, J.G.; Donner, D.B.; Pfeffer, L.M. Interferon alpha /beta promotes cell survival by activating nuclear factor kappa B through phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 2001, 276, 13756–13761. [Google Scholar] [PubMed]
- Kaur, S.; Sassano, A.; Dolniak, B.; Joshi, S.; Majchrzak-Kita, B.; Baker, D.P.; Hay, N.; Fish, E.N.; Platanias, L. C. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 2008, 105, 4808–4813. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef]
- de Veer, M.J.; Holko, M.; Frevel, M.; Walker, E.; Der, S.; Paranjape, J.M.; Silverman, R.H.; Williams, B.R. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 2001, 69, 912–920. [Google Scholar] [PubMed]
- Lanford, R.E.; Guerra, B.; Lee, H.; Chavez, D.; Brasky, K.M.; Bigger, C.B. Genomic response to interferon-alpha in chimpanzees: implications of rapid downregulation for hepatitis C kinetics. Hepatology 2006, 43, 961–972. [Google Scholar] [CrossRef] [PubMed]
- van Boxel-Dezaire, A.H.; Rani, M.R.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Terenzi, F.; Hui, D.J.; Merrick, W.C.; Sen, G.C. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J. Biol. Chem. 2006, 281, 34064–34071. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, J.; Potter, J.L.; Haas, A.L. Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J. Biol. Chem. 1996, 271, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef]
- Giannakopoulos, N.V.; Luo, J.K.; Papov, V.; Zou, W.; Lenschow, D.J.; Jacobs, B.S.; Borden, E.C.; Li, J.; Virgin, H.W.; Zhang, D.E. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem. Biophys. Res. Commun. 2005, 336, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. Embo J. 2001, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.I.; Giannakopoulos, N.V.; Virgin, H.W.; Zhang, D.E. Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol. Cell. Biol. 2004, 24, 9592–9600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Beaudenon, S.L.; Kelley, M.L.; Waddell, M.B.; Yuan, W.; Schulman, B.A.; Huibregtse, J.M.; Krug, R.M. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc. Natl. Acad. Sci. USA 2004, 101, 7578–7582. [Google Scholar] [CrossRef]
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Pung, Y.F.; Sze, N.S.; Chin, K.C. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. USA 2006, 103, 10735–10740. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; Leib, D.A.; Pekosz, A.; Knobeloch, K.P.; Horak, I.; Virgin, H.W.t. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef]
- Helbig, K.J.; Lau, D.T.; Semendric, L.; Harley, H.A.; Beard, M.R. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology 2005, 42, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Gale Jr., M.J.; Korth, M.J.; Tang, N.M.; Tan, S.L.; Hopkins, D.A.; Dever, T.E.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Sakuma, I.; Asahina, Y.; Kurosaki, M.; Murakami, T.; Yamamoto, C.; Ogura, Y.; Izumi, N.; Marumo, F.; Sato, C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 1996, 334, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 1999, 285, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Abid, K.; Quadri, R.; Negro, F. Hepatitis C virus, the E2 envelope protein, and alpha-interferon resistance. Science 2000, 287, 1555. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Heim, M.H. HCV Innate Immune Responses. Viruses 2009, 1, 1073-1088. https://doi.org/10.3390/v1031073
Heim MH. HCV Innate Immune Responses. Viruses. 2009; 1(3):1073-1088. https://doi.org/10.3390/v1031073
Chicago/Turabian StyleHeim, Markus H. 2009. "HCV Innate Immune Responses" Viruses 1, no. 3: 1073-1088. https://doi.org/10.3390/v1031073
APA StyleHeim, M. H. (2009). HCV Innate Immune Responses. Viruses, 1(3), 1073-1088. https://doi.org/10.3390/v1031073