Herpes Virus Amplicon Vectors

Abstract
:1. Introduction
1.1. Overview of Herpes Simplex Virus
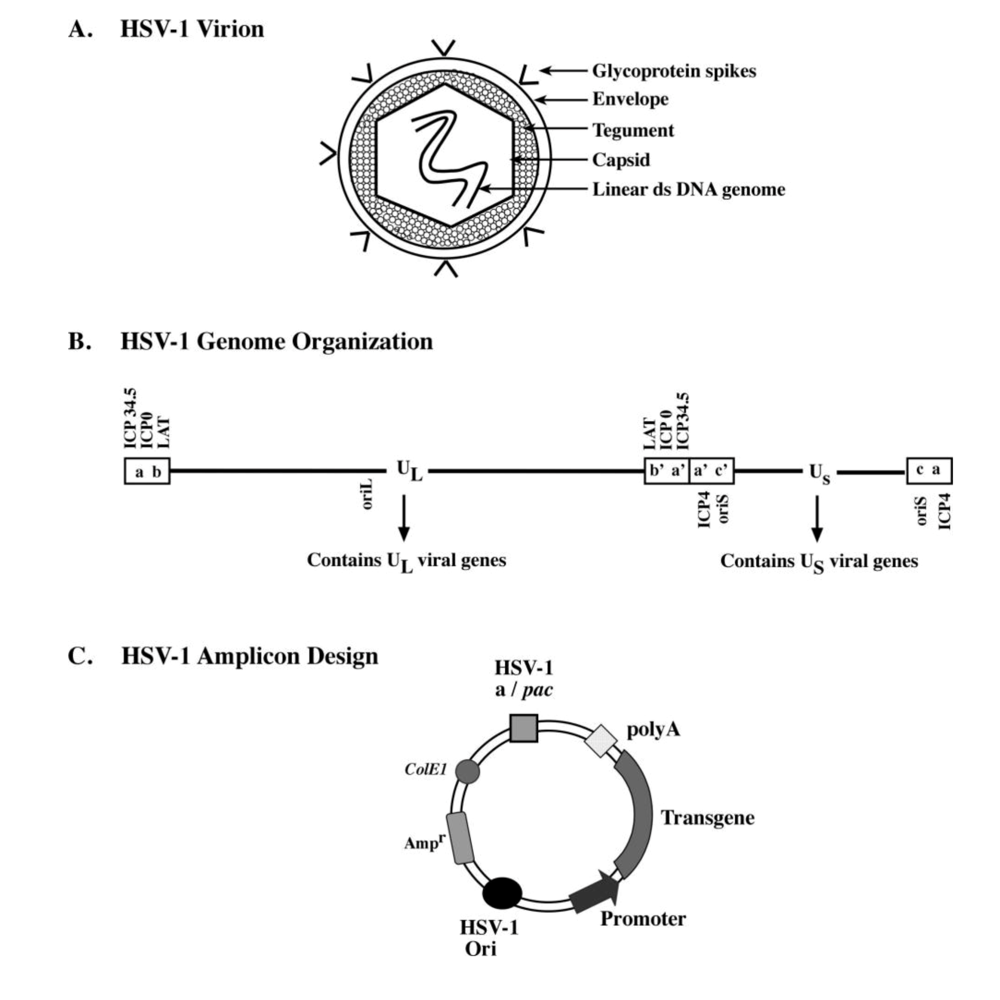
1.1.1. HSV-1 Structure

1.1.2. Virus entry and delivery of genetic payload
1.1.3. Lytic vs. latent infection
1.2. HSV-1 vector generation
2. HSV-1 Amplicon Vectors
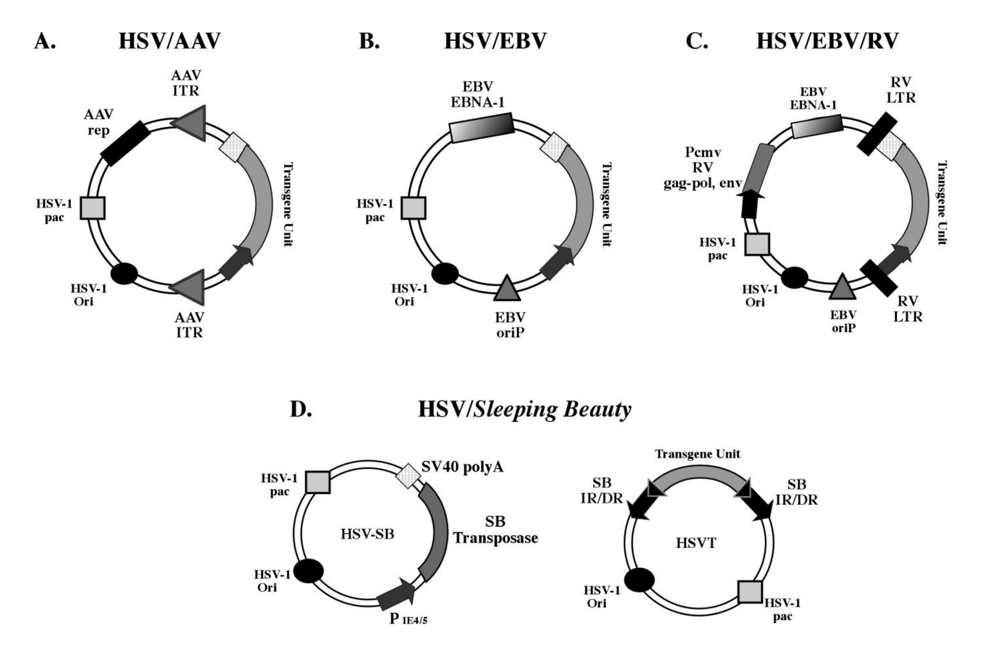
2.1. Types of HSV-1 amplicon vectors

2.1.1. Conventional amplicons
2.1.2. Episomal amplicons
2.1.3. Integration-competent amplicons
2.2. HSV-1 amplicon vector packaging systems
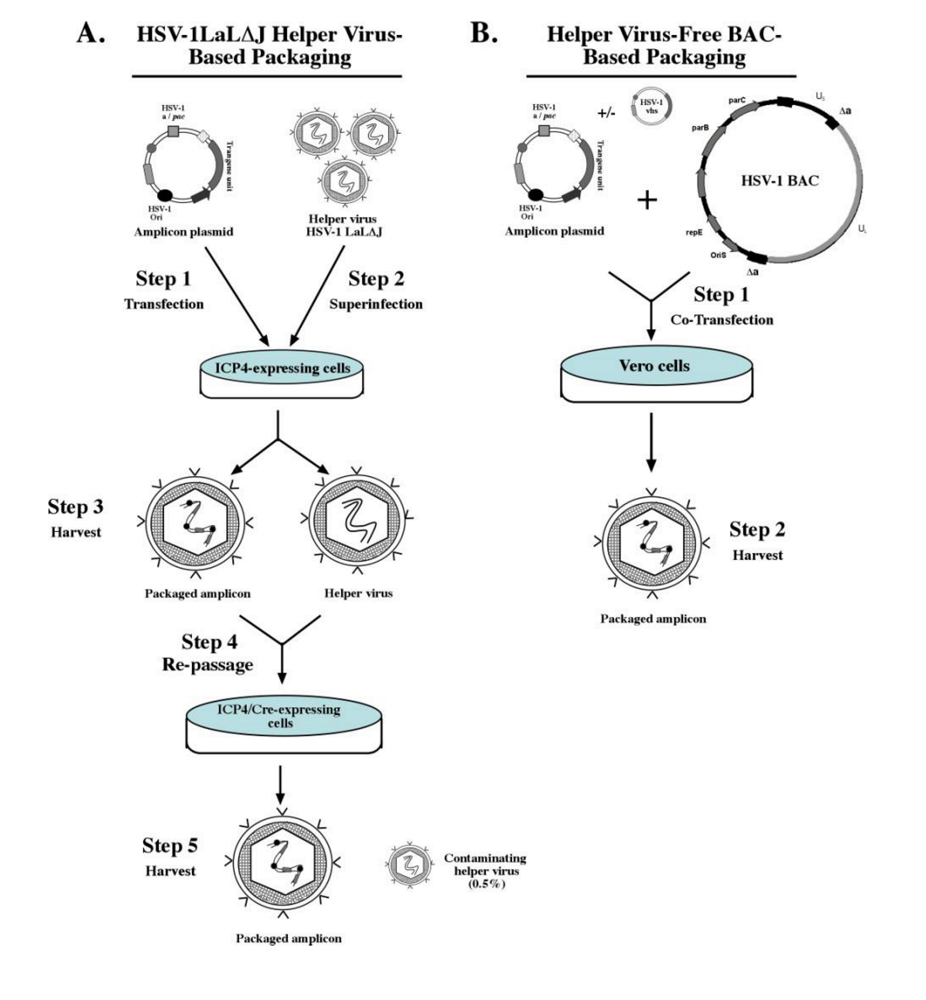
2.2.1. Helper virus-based packaging system
2.2.2. Helper virus-free packaging systems

2.3. HSV-1 virion engineering for targeted transduction
2.4. Immune responses to HSV-1 amplicons
2.5. HSV-1 amplicons for gene therapy
2.5.1. Parkinson’s disease

{kind=link}
{kind=link}
{kind=link}
| Amplicon | Type | Episomal /Integration-Competent | Disease Application | Transgene(s) Delivered(** indicates delivery of entire genomic locus) | References |
|---|---|---|---|---|---|
| HSV-1 | Conventional | Episomal | Parkinson’s disease | GDNFBDNFAADCTHGTP CH1VMAT -2 | [103] [103] [104] [104] [105] [105] |
| Ischemia | GDNFbcl-2GPXHSP72SOD-1GLUT1Calbindin d28k | [106] [107] [108] [109] [110] [111] [112] | |||
| HIV | gp120 | [113] [114] | |||
| Alzheimer’s disease | Amyloid-β (1-42)IL-4 | [115] [116] | |||
| Glioblastoma | Thymidine kinaseTRAILsiRNA α-EGFRGM-CSF | [117] [118] [119] [120] | |||
| Prostate cancer | Prostate-specific antigen (PSA) | [121] | |||
| Chronic Lymphocytic Leukemia | CD80 (B7.1)CD154 (CD40L)SLCCD40L | [122] [123] | |||
| Melanoma | GM-CSF | [124] | |||
| Friedreich’s Ataxia | FRDA cDNA | [125] | |||
| Ataxia Telangiectasia | ATM cDNA | [126] [127,128] | |||
| HSV/EBV | Hybrid | Episomal | Friedreich’s Ataxia | FRDA** | [129] |
| Glioblastoma | ASIC2α | [130] | |||
| Lesch-Nyhan syndrome | HPRT** | [131] | |||
| Familial hypercholesterolemia | LDLR** | [49] | |||
| HSV-BAC-S/MAR | Hybrid | Episomal | Familial hypercholesterolemia | LDLR** | [50] |
| HSV HAC | Hybrid | Episomal | Lesch-Nyhan syndrome | HPRT** | [51] |
| HSV/EBV/RV | Tribrid | Episomal | Glioblastoma | RV gag-pol, env genes | [60] [62] |
| HSV/AAV/RV | Tribrid | Integration-competent | Glioblastoma | RV gag-pol, env genes | [61] |
| HSV/AAV | Hybrid | Integration-competent | Ataxia Telangiectasia | ATM cDNA | [132] |
| Glioblastoma | β-galactosidase | [53] | |||
| Lysosomal storage disease | Human β-galactosidase** | [57] | |||
| HSV/Sleeping Beauty | Hybrid | Integration-competent | In utero delivery | β-galactosidase-neomycin phosphotransferase | [63] [64] |
2.5.2. Ischemia
2.5.3. Cancer therapy
2.5.4. Hereditary ataxia
2.6. Use of HSV-1 amplicons for vaccine development
2.6.1. Cancer vaccines
2.6.2. HIV vaccines
2.6.3. Alzheimer’s disease vaccines
2.7. Current impediments to clinical implementation of HSV-1 amplicons
2.7.1. Scale-up challenges
2.7.2. Extension of gene expression duration
2.7.3. Regulated transgene expression
3. Conclusions
References and Notes
- Spaete, R.R.; Frenkel, N. The herpes simplex virus amplicon: a new eucaryotic defective-virus cloning-amplifying vector. Cell 1982, 30, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, K.; Prichard, M.N.; Duke, G.M.; Kemble, G.W.; Spaete, R.R. Human cytomegalovirus plasmid-based amplicon vector system for gene therapy. Genet. Vaccines Ther. 2005, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.E.; Wade-Martins, R.; James, M.R. Infectious delivery of 120-kilobase genomic DNA by an epstein-barr virus amplicon vector. Mol. Ther. 2002, 5, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Furlong, D.; Swift, H.; Roizman, B. Arrangement of herpesvirus deoxyribonucleic acid in the core. J. Virol. 1972, 10, 1071–1074. [Google Scholar] [PubMed]
- Perry, L.J.; McGeoch, D.J. The DNA sequences of the long repeat region and adjoining parts of the long unique region in the genome of herpes simplex virus type 1 . J. Gen. Virol. 1988, 69 ( Pt 11), 2831–2846. [Google Scholar] [CrossRef] [PubMed]
- Deiss, L.P.; Chou, J.; Frenkel, N. Functional domains within the a sequence involved in the cleavage-packaging of herpes simplex virus DNA. J. Virol. 1986, 59, 605–618. [Google Scholar] [PubMed]
- Vlazny, D.A.; Frenkel, N. Replication of herpes simplex virus DNA: localization of replication recognition signals within defective virus genomes. Proc. Natl. Acad. Sci. U S A 1981, 78, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Krisky, D.M.; Wolfe, D.; Goins, W.F.; Marconi, P.C.; Ramakrishnan, R.; Mata, M.; Rouse, R.J.; Fink, D.J.; Glorioso, J.C. Deletion of multiple immediate-early genes from herpes simplex virus reduces cytotoxicity and permits long-term gene expression in neurons. Gene Ther. 1998, 5, 1593–1603. [Google Scholar] [PubMed]
- Zhou, Z.H.; Dougherty, M.; Jakana, J.; He, J.; Rixon, F.J.; Chiu, W. Seeing the herpesvirus capsid at 8.5 A . Science 2000, 288, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Cardone, G.; Winkler, D.C.; Trus, B.L.; Cheng, N.; Heuser, J.E.; Newcomb, W.W.; Brown, J.C.; Steven, A.C. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 2007, 361, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, W.W.; Juhas, R.M.; Thomsen, D.R.; Homa, F.L.; Burch, A.D.; Weller, S.K.; Brown, J.C. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 2001, 75, 10923–10932. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Duffy, C.; Mbong, E.F.; Baines, J.D. VP22 of herpes simplex virus 1 promotes protein synthesis at late times in infection and accumulation of a subset of viral mRNAs at early times in infection. J. Virol. 2009, 83, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; Guay, G.; Lippe, R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef] [PubMed]
- Laquerre, S.; Argnani, R.; Anderson, D.B.; Zucchini, S.; Manservigi, R.; Glorioso, J.C. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 1998, 72, 6119–6130. [Google Scholar] [PubMed]
- Pertel, P.E.; Fridberg, A.; Parish, M.L.; Spear, P.G. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 2001, 279, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.A.; Fink, D.J.; Glorioso, J.C. Gene delivery using herpes simplex virus vectors. DNA Cell Biol. 2002, 21, 915–936. [Google Scholar] [PubMed]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B . J. Gen. Virol. 1994, 75(Pt 6), 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [PubMed]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; Spear, P.G.; Lanier, L.L.; Arase, H. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.S.; Geraghty, R.J.; Martinez, W.M.; Montgomery, R.I.; Whitbeck, J.C.; Xu, R.; Eisenberg, R.J.; Cohen, G.H.; Spear, P.G. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 1998, 246, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Horikawa, K.; Nakanishi, H.; Takahashi, K.; Miyahara, M.; Nishimura, M.; Tachibana, K.; Mizoguchi, A.; Takai, Y. Nectin-3, a new member of immunoglobulin-like cell adhesion molecules that shows homophilic and heterophilic cell-cell adhesion activities. J. Biol. Chem. 2000, 275, 10291–10299. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.; Bruun, B.; Minson, T.; Browne, H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 1998, 72, 873–875. [Google Scholar] [PubMed]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Ojala, P.M.; Sodeik, B.; Ebersold, M.W.; Kutay, U.; Helenius, A. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell Biol. 2000, 20, 4922–4931. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.A.; DeLuca, N.A. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. U S A 2003, 100, 7871–7876. [Google Scholar] [CrossRef] [PubMed]
- Garber, D.A.; Beverley, S.M.; Coen, D.M. Demonstration of circularization of herpes simplex virus DNA following infection using pulsed field gel electrophoresis. Virology 1993, 197, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Mellerick, D.M.; Fraser, N.W. Physical state of the latent herpes simplex virus genome in a mouse model system: evidence suggesting an episomal state. Virology 1987, 158, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [PubMed]
- DeLuca, N.A.; Schaffer, P.A. Activation of immediate-early, early, and late promoters by temperature-sensitive and wild-type forms of herpes simplex virus type 1 protein ICP4. Mol. Cell Biol. 1985, 5, 1997–2008. [Google Scholar] [PubMed]
- Sacks, W.R.; Greene, C.C.; Aschman, D.P.; Schaffer, P.A. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 1985, 55, 796–805. [Google Scholar] [PubMed]
- Nsiah, Y.A.; Rapp, F. Role of latency-associated transcript in herpes simplex virus infection. Intervirology 1991, 32, 101–115. [Google Scholar] [PubMed]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Gartner, J.J.; Sethupathy, P.; Hatzigeorgiou, A.G.; Fraser, N.W. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature 2006, 442, 82–85. [Google Scholar] [PubMed]
- Burton, E.A.; Fink, D.J.; Glorioso, J.C. Replication-defective genomic HSV Gene Ther.apy vectors: design, production and CNS applications. Curr. Opin. Mol. Ther. 2005, 7, 326–336. [Google Scholar] [PubMed]
- Boviatsis, E.J.; Scharf, J.M.; Chase, M.; Harrington, K.; Kowall, N.W.; Breakefield, X.O.; Chiocca, E.A. Antitumor activity and reporter gene transfer into rat brain neoplasms inoculated with herpes simplex virus vectors defective in thymidine kinase or ribonucleotide reductase. Gene Ther. 1994, 1, 323–331. [Google Scholar] [PubMed]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; Martuza, R.L. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef]
- DeLuca, N.A.; McCarthy, A.M.; Schaffer, P.A. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 1985, 56, 558–570. [Google Scholar] [PubMed]
- Samaniego, L.A.; Webb, A.L.; DeLuca, N.A. Functional interactions between herpes simplex virus immediate-early proteins during infection: gene expression as a consequence of ICP27 and different domains of ICP4. J. Virol. 1995, 69, 5705–5715. [Google Scholar] [PubMed]
- Assudani, D.P.; Ahmad, M.; Li, G.; Rees, R.C.; Ali, S.A. Immunotherapeutic potential of DISC-HSV and OX40L in cancer. Cancer Immunol. Immunother. 2006, 55, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Oehmig, A.; Fraefel, C.; Breakefield, X.O. Update on herpesvirus amplicon vectors. Mol. Ther. 2004, 10, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Lufino, M.M.; Edser, P.A.; Wade-Martins, R. Advances in high-capacity extrachromosomal vector technology: episomal maintenance, vector delivery, and transgene expression. Mol. Ther. 2008, 16, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Vos, J.M. A hybrid herpesvirus infectious vector based on Epstein-Barr virus and herpes simplex virus type 1 for gene transfer into human cells in vitro and in vivo. J. Virol. 1996, 70, 8422–8430. [Google Scholar] [PubMed]
- Wade-Martins, R.; White, R.E.; Kimura, H.; Cook, P.R.; James, M.R. Stable correction of a genetic deficiency in human cells by an episome carrying a 115 kb genomic transgene. Nat. Biotechnol. 2000, 18, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Wade-Martins, R.; Saeki, Y.; Chiocca, E.A. Infectious delivery of a 135-kb LDLR genomic locus leads to regulated complementation of low-density lipoprotein receptor deficiency in human cells. Mol. Ther. 2003, 7, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Lufino, M.M.; Manservigi, R.; Wade-Martins, R. An S/MAR-based infectious episomal genomic DNA expression vector provides long-term regulated functional complementation of LDLR deficiency . Nucleic Acids Res. 2007, 35, e98. [Google Scholar] [CrossRef] [PubMed]
- Moralli, D.; Simpson, K.M.; Wade-Martins, R.; Monaco, Z.L. A novel human artificial chromosome gene expression system using herpes simplex virus type 1 vectors. EMBO Rep. 2006, 7, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Oehmig, A.; Fraefel, C.; Breakefield, X.O.; Ackermann, M. Herpes simplex virus type 1 amplicons and their hybrid virus partners, EBV, AAV, and retrovirus. Curr. Gene Ther. 2004, 4, 385–408. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.M.; Jacoby, D.; Pechan, P.A.; Fraefel, C.; Borghesani, P.; Schuback, D.; Dunn, R.J.; Smith, F.I.; Breakefield, X.O. HSV/AAV hybrid amplicon vectors extend transgene expression in human glioma cells. Hum. Gene Ther. 1997, 8, 359–370. [Google Scholar] [CrossRef]
- Glauser, D.L.; Ackermann, M.; Saydam, O.; Fraefel, C. Chimeric herpes simplex virus/adeno-associated virus amplicon vectors. Curr. Gene Ther. 2006, 6, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Costantini, L.C.; Jacoby, D.R.; Wang, S.; Fraefel, C.; Breakefield, X.O.; Isacson, O. Gene transfer to the nigrostriatal system by hybrid herpes simplex virus/adeno-associated virus amplicon vectors. Hum. Gene Ther. 1999, 10, 2481–2494. [Google Scholar] [PubMed]
- Fraefel, C.; Jacoby, D.R.; Lage, C.; Hilderbrand, H.; Chou, J.Y.; Alt, F.W.; Breakefield, X.O.; Majzoub, J.A. Gene transfer into hepatocytes mediated by helper virus-free HSV/AAV hybrid vectors. Mol. Med. 1997, 3, 813–825. [Google Scholar] [PubMed]
- Oehmig, A.; Cortes, M.L.; Perry, K.F.; Sena-Esteves, M.; Fraefel, C.; Breakefield, X.O. Integration of active human beta-galactosidase gene (100 kb) into genome using HSV/AAV amplicon vector. Gene Ther. 2007, 14, 1078–1091. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, R.; Burkle, A.; Stephan, S.; zur Hausen, H. The adeno-associated virus rep gene suppresses herpes simplex virus-induced DNA amplification. J. Virol. 1990, 64, 3012–3018. [Google Scholar] [PubMed]
- Liu, Q.; Perez, C.F.; Wang, Y. Efficient site-specific integration of large transgenes by an enhanced herpes simplex virus/adeno-associated virus hybrid amplicon vector. J. Virol. 2006, 80, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Sena-Esteves, M.; Hampl, J.A.; Camp, S.M.; Breakefield, X.O. Generation of stable retrovirus packaging cell lines after transduction with herpes simplex virus hybrid amplicon vectors. J. Gene Med. 2002, 4, 229–239. [Google Scholar] [CrossRef]
- Sena-Esteves, M.; Saeki, Y.; Camp, S.M.; Chiocca, E.A.; Breakefield, X.O. Single-step conversion of cells to retrovirus vector producers with herpes simplex virus-Epstein-Barr virus hybrid amplicons. J. Virol. 1999, 73, 10426–10439. [Google Scholar] [PubMed]
- Hampl, J.A.; Camp, S.M.; Mydlarz, W.K.; Hampl, M.; Ichikawa, T.; Chiocca, E.A.; Louis, D.N.; Sena-Esteves, M.; Breakefield, X.O. Potentiated gene delivery to tumors using herpes simplex virus/Epstein-Barr virus/RV tribrid amplicon vectors. Hum. Gene Ther. 2003, 14, 611–626. [Google Scholar] [PubMed]
- Bowers, W.J.; Mastrangelo, M.A.; Howard, D.F.; Southerland, H.A.; Maguire-Zeiss, K.A.; Federoff, H.J. Neuronal precursor-restricted transduction via in utero CNS gene delivery of a novel bipartite HSV amplicon/transposase hybrid vector. Mol. Ther. 2006, 13, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.B.; Mastrangelo, M.A.; Federoff, H.J.; Bowers, W.J. Neuronal specificity of HSV/sleeping beauty amplicon transduction in utero is driven primarily by tropism and cell type composition. Mol. Ther. 2007, 15, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.I.; Keyomarsi, K.; Bryan, J.; Pardee, A.B. An efficient deletion mutant packaging system for defective herpes simplex virus vectors: potential applications to human Gene Ther.apy and neuronal physiology . Proc. Natl. Acad. Sci. U S A 1990, 87, 8950–8954. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.; Hartley, D.; Starr, P.; Lang, P.; Song, S.; Yu, L.; Wang, Y.; Geller, A.I. Generation of high-titer defective HSV-1 vectors using an IE 2 deletion mutant and quantitative study of expression in cultured cortical cells. Biotechniques 1996, 20, 460–469. [Google Scholar] [PubMed]
- Olschowka, J.A.; Bowers, W.J.; Hurley, S.D.; Mastrangelo, M.A.; Federoff, H.J. Helper-free HSV-1 amplicons elicit a markedly less robust innate immune response in the CNS. Mol. Ther. 2003, 7, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.L. HSV-1-derived amplicon vectors: recent technological improvements and remaining difficulties--a review. Mem. Inst. Oswaldo Cruz 2009, 104, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Logvinoff, C.; Epstein, A.L. A novel approach for herpes simplex virus type 1 amplicon vector production, using the Cre-loxP recombination system to remove helper virus. Hum. Gene Ther. 2001, 12, 161–167. [Google Scholar] [PubMed]
- Zaupa, C.; Revol-Guyot, V.; Epstein, A.L. Improved packaging system for generation of high-level noncytotoxic HSV-1 amplicon vectors using Cre-loxP site-specific recombination to delete the packaging signals of defective helper genomes. Hum. Gene Ther. 2003, 14, 1049–1063. [Google Scholar] [PubMed]
- Halterman, M.W.; Giuliano, R.E.; Bowers, W.J.; Federoff, H.J. Improved HSV-1 amplicon packaging using virion host shutoff mutants lacking mRNAse activity. J. Gene Med. 2006, 8, 1320–1328. [Google Scholar] [CrossRef]
- Cunningham, C.; Davison, A.J. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 1993, 197, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Fraefel, C.; Song, S.; Lim, F.; Lang, P.; Yu, L.; Wang, Y.; Wild, P.; Geller, A.I. Helper virus-free transfer of herpes simplex virus type 1 plasmid vectors into neural cells. J. Virol. 1996, 70, 7190–7197. [Google Scholar] [PubMed]
- Saeki, Y.; Ichikawa, T.; Saeki, A.; Chiocca, E.A.; Tobler, K.; Ackermann, M.; Breakefield, X.O.; Fraefel, C. Herpes simplex virus type 1 DNA amplified as bacterial artificial chromosome in Escherichia coli: rescue of replication-competent virus progeny and packaging of amplicon vectors. Hum. Gene Ther. 1998, 9, 2787–2794. [Google Scholar] [CrossRef]
- Saeki, Y.; Fraefel, C.; Ichikawa, T.; Breakefield, X.O.; Chiocca, E.A. Improved helper virus-free packaging system for HSV amplicon vectors using an ICP27-deleted, oversized HSV-1 DNA in a bacterial artificial chromosome. Mol. Ther. 2001, 3, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, T.A.; Strathdee, C.A. An enhanced packaging system for helper-dependent herpes simplex virus vectors. J. Virol. 1998, 72, 7137–7143. [Google Scholar] [PubMed]
- Bowers, W.J.; Howard, D.F.; Brooks, A.I.; Halterman, M.W.; Federoff, H.J. Expression of vhs and VP16 during HSV-1 helper virus-free amplicon packaging enhances titers. Gene Ther. 2001, 8, 111–120. [Google Scholar] [CrossRef]
- Laquerre, S.; Anderson, D.B.; Stolz, D.B.; Glorioso, J.C. Recombinant herpes simplex virus type 1 engineered for targeted binding to erythropoietin receptor-bearing cells. J. Virol. 1998, 72, 9683–9697. [Google Scholar] [PubMed]
- Argnani, R.; Boccafogli, L.; Marconi, P.C.; Manservigi, R. Specific targeted binding of herpes simplex virus type 1 to hepatocytes via the human hepatitis B virus preS1 peptide. Gene Ther. 2004, 11, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.B.; Laquerre, S.; Ghosh, K.; Ghosh, H.P.; Goins, W.F.; Cohen, J.B.; Glorioso, J.C. Pseudotyping of glycoprotein D-deficient herpes simplex virus type 1 with vesicular stomatitis virus glycoprotein G enables mutant virus attachment and entry. J. Virol. 2000, 74, 2481–2487. [Google Scholar] [CrossRef] [PubMed]
- Grandi, P.; Wang, S.; Schuback, D.; Krasnykh, V.; Spear, M.; Curiel, D.T.; Manservigi, R.; Breakefield, X.O. HSV-1 virions engineered for specific binding to cell surface receptors. Mol. Ther. 2004, 9, 419–427. [Google Scholar] [CrossRef]
- Wang, X.; Kong, L.; Zhang, G.R.; Sun, M.; Geller, A.I. Targeted gene transfer to nigrostriatal neurons in the rat brain by helper virus-free HSV-1 vector particles that contain either a chimeric HSV-1 glycoprotein C-GDNF or a gC-BDNF protein. Brain Res. Mol. Brain Res. 2005, 139, 88–102. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, G.R.; Wang, X.; Kong, L.; Geller, A.I. Enhanced nigrostriatal neuron-specific, long-term expression by using neural-specific promoters in combination with targeted gene transfer by modified helper virus-free HSV-1 vector particles. BMC Neurosci. 2008, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Bowers, W.J.; Olschowka, J.A.; Federoff, H.J. Immune responses to replication-defective HSV-1 type vectors within the CNS: implications for Gene Ther.apy . Gene Ther. 2003, 10, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.A.; Federoff, H.J. Immune Responses to Herpes Viral Vectors . Hum. Gene Ther. 2009. [Google Scholar]
- Rasmussen, S.B.; Reinert, L.S.; Paludan, S.R. Innate recognition of intracellular pathogens: detection and activation of the first line of defense. Apmis 2009, 117, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Parsy, M.L.; Orr, A. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J. Virol. 2009, 83, 4963–4977. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Song, B.; Knipe, D.M. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 2008, 374, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Favoreel, H.W.; Nauwynck, H.J.; Pensaert, M.B. Immunological hiding of herpesvirus-infected cells. Arch. Virol. 2000, 145, 1269–1290. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Spear, P.G. Infections with herpes simplex viruses (1). N. Engl. J. Med. 1986, 314, 686–691. [Google Scholar] [PubMed]
- Frank, I.; Friedman, H.M. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J. Virol. 1989, 63, 4479–4488. [Google Scholar] [PubMed]
- Hill, A.B.; Barnett, B.C.; McMichael, A.J.; McGeoch, D.J. HLA class I molecules are not transported to the cell surface in cells infected with herpes simplex virus types 1 and 2. J. Immunol. 1994, 152, 2736–2741. [Google Scholar] [PubMed]
- Tomazin, R.; Hill, A.B.; Jugovic, P.; York, I.; van Endert, P.; Ploegh, H.L.; Andrews, D.W.; Johnson, D.C. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. Embo J. 1996, 15, 3256–3266. [Google Scholar] [PubMed]
- Wood, M.J.; Byrnes, A.P.; Pfaff, D.W.; Rabkin, S.D.; Charlton, H.M. Inflammatory effects of gene transfer into the CNS with defective HSV-1 vectors. Gene Ther. 1994, 1, 283–291. [Google Scholar] [PubMed]
- Tsitoura, E.; Thomas, J.; Cuchet, D.; Thoinet, K.; Mavromara, P.; Epstein, A.L. Infection with herpes simplex type 1-based amplicon vectors results in an IRF3/7-dependent, TLR-independent activation of the innate antiviral response in primary human fibroblasts. J. Gen. Virol. 2009, 90, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kasai, K.; Saeki, Y. Plasmid DNA sequences present in conventional herpes simplex virus amplicon vectors cause rapid transgene silencing by forming inactive chromatin. J. Virol. 2006, 80, 3293–3300. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Chiocca, E.A.; Saeki, Y. Early STAT1 activation after systemic delivery of HSV amplicon vectors suppresses transcription of the vector-encoded transgene. Mol. Ther. 2007, 15, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kasai, K.; Ohtsuki, A.; Godlewski, J.; Nowicki, M.O.; Chiocca, E.A.; Saeki, Y. ICP0 inhibits the decrease of HSV amplicon-mediated transgene expression. Mol. Ther. 2009, 17, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Eidson, K.M.; Hobbs, W.E.; Manning, B.J.; Carlson, P.; DeLuca, N.A. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 2002, 76, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Chiocca, E.A.; Saeki, Y. Stable transgene expression from HSV amplicon vectors in the brain: potential involvement of immunoregulatory signals. Mol. Ther. 2008, 16, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, K.J.; Mendoza, M.R.; Yahr, M.D. Parkinson's disease and long-term levodopa therapy. Adv. Neurol. 1987, 45, 463–467. [Google Scholar] [PubMed]
- Tashiro, Y.; Kaneko, T.; Sugimoto, T.; Nagatsu, I.; Kikuchi, H.; Mizuno, N. Striatal neurons with aromatic L-amino acid decarboxylase-like immunoreactivity in the rat. Neurosci. Lett. 1989, 100, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kong, L.; Wang, X.; Lu, X.G.; Gao, Q.; Geller, A.I. Comparison of the capability of GDNF, BDNF, or both, to protect nigrostriatal neurons in a rat model of Parkinson's disease. Brain Res. 2005, 1052, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, G.R.; Kong, L.; Holmes, C.; Wang, X.; Zhang, W.; Goldstein, D.S.; Geller, A.I. Correction of a rat model of Parkinson's disease by coexpression of tyrosine hydroxylase and aromatic amino acid decarboxylase from a helper virus-free herpes simplex virus type 1 vector. Hum. Gene Ther. 2003, 14, 415–424. [Google Scholar] [PubMed]
- Sun, M.; Kong, L.; Wang, X.; Holmes, C.; Gao, Q.; Zhang, G.R.; Pfeilschifter, J.; Goldstein, D.S.; Geller, A.I. Coexpression of tyrosine hydroxylase, GTP cyclohydrolase I, aromatic amino acid decarboxylase, and vesicular monoamine transporter 2 from a helper virus-free herpes simplex virus type 1 vector supports high-level, long-term biochemical and behavioral correction of a rat model of Parkinson's disease. Hum. Gene Ther. 2004, 15, 1177–1196. [Google Scholar] [PubMed]
- Harvey, B.K.; Chang, C.F.; Chiang, Y.H.; Bowers, W.J.; Morales, M.; Hoffer, B.J.; Wang, Y.; Federoff, H.J. HSV amplicon delivery of glial cell line-derived neurotrophic factor is neuroprotective against ischemic injury. Exp. Neurol. 2003, 183, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Antonawich, F.J.; Federoff, H.J.; Davis, J.N. BCL-2 transduction, using a herpes simplex virus amplicon, protects hippocampal neurons from transient global ischemia. Exp. Neurol. 1999, 156, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, B.; Yenari, M.A.; Sapolsky, R.M.; Steinberg, G.K. Glutathione peroxidase overexpression inhibits cytochrome C release and proapoptotic mediators to protect neurons from experimental stroke. Stroke 2003, 34, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Fink, S.L.; Sun, G.H.; Chang, L.K.; Patel, M.K.; Kunis, D.M.; Onley, D.; Ho, D.Y.; Sapolsky, R.M.; Steinberg, G.K. Gene Ther.apy with HSP72 is neuroprotective in rat models of stroke and epilepsy . Ann. Neurol. 1998, 44, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.S.; Zhao, H.; Sun, G.H.; Sapolsky, R.M.; Steinberg, G.K. Gene Ther.apy using SOD1 protects striatal neurons from experimental stroke . Neurosci. Lett. 2007, 411, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Ho, D.Y.; Dash, R.; Sapolsky, R.M. Herpes simplex virus vectors overexpressing the glucose transporter gene protect against seizure-induced neuron loss. Proc. Natl. Acad. Sci. U S A 1995, 92, 7247–7251. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Minami, M.; Sun, G.H.; Meier, T.J.; Kunis, D.M.; McLaughlin, J.R.; Ho, D.Y.; Sapolsky, R.M.; Steinberg, G.K. Calbindin d28k overexpression protects striatal neurons from transient focal cerebral ischemia. Stroke 2001, 32, 1028–1035. [Google Scholar] [PubMed]
- Gorantla, S.; Santos, K.; Meyer, V.; Dewhurst, S.; Bowers, W.J.; Federoff, H.J.; Gendelman, H.E.; Poluektova, L. Human dendritic cells transduced with herpes simplex virus amplicons encoding human immunodeficiency virus type 1 (HIV-1) gp120 elicit adaptive immune responses from human cells engrafted into NOD/SCID mice and confer partial protection against HIV-1 challenge. J. Virol. 2005, 79, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wiley, R.D.; Evans, T.G.; Bowers, W.J.; Federoff, H.J.; Dewhurst, S. Cellular immune responses to helper-free HSV-1 amplicon particles encoding HIV-1 gp120 are enhanced by DNA priming. Vaccine 2003, 21, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Bowers, W.J.; Mastrangelo, M.A.; Stanley, H.A.; Casey, A.E.; Milo, L.J.; Federoff, H.J. HSV amplicon-mediated Abeta vaccination in Tg2576 mice: differential antigen-specific immune responses . Neurobiol. Aging 2005, 26, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Frazer, M.E.; Hughes, J.E.; Mastrangelo, M.A.; Tibbens, J.L.; Federoff, H.J.; Bowers, W.J. Reduced pathology and improved behavioral performance in Alzheimer's disease mice vaccinated with HSV amplicons expressing amyloid-beta and interleukin-4. Mol. Ther. 2008, 16, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qi, J.; Smith, M.; Link, C.J. Antitumor effects on human melanoma xenografts of an amplicon vector transducing the herpes thymidine kinase gene followed by ganciclovir. Cancer Gene Ther. 2002, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Tang, Y.; Breakefield, X.; Weissleder, R. Real-time imaging of TRAIL-induced apoptosis of glioma tumors in vivo. Oncogene 2003, 22, 6865–6872. [Google Scholar] [CrossRef] [PubMed]
- Saydam, O.; Glauser, D.L.; Heid, I.; Turkeri, G.; Hilbe, M.; Jacobs, A.H.; Ackermann, M.; Fraefel, C. Herpes simplex virus 1 amplicon vector-mediated siRNA targeting epidermal growth factor receptor inhibits growth of human glioma cells in vivo. Mol. Ther. 2005, 12, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Herrlinger, U.; Jacobs, A.; Quinones, A.; Woiciechowsky, C.; Sena-Esteves, M.; Rainov, N.G.; Fraefel, C.; Breakefield, X.O. Helper virus-free herpes simplex virus type 1 amplicon vectors for granulocyte-macrophage colony-stimulating factor-enhanced vaccination therapy for experimental glioma. Hum Gene Ther. 2000, 11, 1429–1438. [Google Scholar] [PubMed]
- Willis, R.A.; Bowers, W.J.; Turner, M.J.; Fisher, T.L.; Abdul-Alim, C.S.; Howard, D.F.; Federoff, H.J.; Lord, E.M.; Frelinger, J.G. Dendritic cells transduced with HSV-1 amplicons expressing prostate-specific antigen generate antitumor immunity in mice. Hum Gene Ther. 2001, 12, 1867–1879. [Google Scholar] [PubMed]
- Tolba, K.A.; Bowers, W.J.; Hilchey, S.P.; Halterman, M.W.; Howard, D.F.; Giuliano, R.E.; Federoff, H.J.; Rosenblatt, J.D. Development of herpes simplex virus-1 amplicon-based immunotherapy for chronic lymphocytic leukemia. Blood 2001, 98, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Tolba, K.A.; Bowers, W.J.; Muller, J.; Housekneckt, V.; Giuliano, R.E.; Federoff, H.J.; Rosenblatt, J.D. Herpes simplex virus (HSV) amplicon-mediated codelivery of secondary lymphoid tissue chemokine and CD40L results in augmented antitumor activity. Cancer Res. 2002, 62, 6545–6551. [Google Scholar] [PubMed]
- Toda, M.; Martuza, R.L.; Rabkin, S.D. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte-macrophage colony-stimulating factor. Mol. Ther. 2000, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.; Palomo, G.M.; Mauritz, C.; Gimenez-Cassina, A.; Illana, B.; Wandosell, F.; Diaz-Nido, J. Functional recovery in a Friedreich's ataxia mouse model by frataxin gene transfer using an HSV-1 amplicon vector. Mol. Ther. 2007, 15, 1072–1078. [Google Scholar] [PubMed]
- Qi, J.; Shackelford, R.; Manuszak, R.; Cheng, D.; Smith, M.; Link, C.J.; Wang, S. Functional expression of ATM gene carried by HSV amplicon vector in vitro and in vivo. Gene Ther. 2004, 11, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.L.; Bakkenist, C.J.; Di Maria, M.V.; Kastan, M.B.; Breakefield, X.O. HSV-1 amplicon vector-mediated expression of ATM cDNA and correction of the ataxia-telangiectasia cellular phenotype. Gene Ther. 2003, 10, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.L.; Oehmig, A.; Perry, K.F.; Sanford, J.D.; Breakefield, X.O. Expression of human ATM cDNA in Atm-deficient mouse brain mediated by HSV-1 amplicon vector. Neuroscience 2006, 141, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sebastian, S.; Gimenez-Cassina, A.; Diaz-Nido, J.; Lim, F.; Wade-Martins, R. Infectious delivery and expression of a 135 kb human FRDA genomic DNA locus complements Friedreich's ataxia deficiency in human cells. Mol. Ther. 2007, 15, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Tannous, B.A.; Christensen, A.P.; Pike, L.; Wurdinger, T.; Perry, K.F.; Saydam, O.; Jacobs, A.H.; Garcia-Anoveros, J.; Weissleder, R.; Sena-Esteves, M.; Corey, D.P.; Breakefield, X.O. Mutant sodium channel for tumor therapy. Mol. Ther. 2009, 17, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Wade-Martins, R.; Smith, E.R.; Tyminski, E.; Chiocca, E.A.; Saeki, Y. An infectious transfer and expression system for genomic DNA loci in human and mouse cells. Nat. Biotechnol. 2001, 19, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.L.; Oehmig, A.; Saydam, O.; Sanford, J.D.; Perry, K.F.; Fraefel, C.; Breakefield, X.O. Targeted integration of functional human ATM cDNA into genome mediated by HSV/AAV hybrid amplicon vector. Mol. Ther. 2008, 16, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Reinblatt, M.; Pin, R.H.; Bowers, W.J.; Federoff, H.J.; Fong, Y. Herpes simplex virus amplicon delivery of a hypoxia-inducible soluble vascular endothelial growth factor receptor (sFlk-1) inhibits angiogenesis and tumor growth in pancreatic adenocarcinoma. Ann. Surg. Oncol. 2005, 12, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Yanoma, S.; Nishimura, G.; Hattori, S.; Ito, T.; Okudera, K.; Tsukuda, M. Therapeutic efficiency of IL-2 gene transduced tumor vaccine for head and neck carcinoma. Cancer Lett. 2000, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Jarnagin, W.R.; Delman, K.; Kooby, D.; Mastorides, S.; Zager, J.; Brennan, M.F.; Blumgart, L.H.; Federoff, H.; Fong, Y. Neoadjuvant interleukin-12 immunoGene Ther.apy protects against cancer recurrence after liver resection in an animal model. Ann. Surg. 2000, 231, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Loew, S.; Schmidt, U.; Unterberg, A.; Halatsch, M.E. The epidermal growth factor receptor as a therapeutic target in glioblastoma multiforme and other malignant neoplasms. Anticancer Agents Med. Chem. 2009, 9, 703–715. [Google Scholar] [PubMed]
- Pechan, P.A.; Herrlinger, U.; Aghi, M.; Jacobs, A.; Breakefield, X.O. Combined HSV-1 recombinant and amplicon piggyback vectors: replication-competent and defective forms, and therapeutic efficacy for experimental gliomas. J. Gene Med. 1999, 1, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Bu, L.X.; Rennie, P.S.; Jia, W.W. An HSV-1 amplicon system for prostate-specific expression of ICP4 to complement oncolytic viral replication for in vitro and in vivo treatment of prostate cancer cells. Cancer Gene Ther. 2007, 14, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Shiloh, Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol 1997, 15, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; Zara, F.; Canizares, J.; Koutnikova, H.; Bidichandani, S.I.; Gellera, C.; Brice, A.; Trouillas, P.; De Michele, G.; Filla, A.; De Frutos, R.; Palau, F.; Patel, P.I.; Di Donato, S.; Mandel, J.L.; Cocozza, S.; Koenig, M.; Pandolfo, M. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [PubMed]
- Hocknell, P.K.; Wiley, R.D.; Wang, X.; Evans, T.G.; Bowers, W.J.; Hanke, T.; Federoff, H.J.; Dewhurst, S. Expression of human immunodeficiency virus type 1 gp120 from herpes simplex virus type 1-derived amplicons results in potent, specific, and durable cellular and humoral immune responses. J. Virol. 2002, 76, 5565–5580. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Janus, C.; Pearson, J.; McLaurin, J.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Chishti, M.A.; Horne, P.; Heslin, D.; French, J.; Mount, H.T.; Nixon, R.A.; Mercken, M.; Bergeron, C.; Fraser, P.E.; St George-Hyslop, P.; Westaway, D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 2000, 408, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Diamond, D.M.; Gottschall, P.E.; Ugen, K.E.; Dickey, C.; Hardy, J.; Duff, K.; Jantzen, P.; DiCarlo, G.; Wilcock, D.; Connor, K.; Hatcher, J.; Hope, C.; Gordon, M.; Arendash, G.W. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 2000, 408, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; Kholodenko, D.; Lee, M.; Liao, Z.; Lieberburg, I.; Motter, R.; Mutter, L.; Soriano, F.; Shopp, G.; Vasquez, N.; Vandevert, C.; Walker, S.; Wogulis, M.; Yednock, T.; Games, D.; Seubert, P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Bowers, W.J.; Federoff, H.J. Amyloid immunotherapy-engendered CNS inflammation . Neurobiol. Aging 2002, 23, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.M.; Gilman, S.; Dartigues, J.F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; Michel, B.F.; Boada, M.; Frank, A.; Hock, C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [PubMed]
- Hock, C.; Konietzko, U.; Papassotiropoulos, A.; Wollmer, A.; Streffer, J.; von Rotz, R.C.; Davey, G.; Moritz, E.; Nitsch, R.M. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat. Med. 2002, 8, 1270–1275. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; Kwong, A.D.; Kleopoulos, S.P.; Mobbs, C.V.; Rabkin, S.D.; Pfaff, D.W. Preproenkephalin promoter yields region-specific and long-term expression in adult brain after direct in vivo gene transfer via a defective herpes simplex viral vector. Proc. Natl. Acad. Sci. U S A 1994, 91, 8979–8983. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.K.; Belloni, M.; Conti, B.; Federoff, H.J.; Starr, R.; Son, J.H.; Baker, H.; Joh, T.H. Prolonged in vivo gene expression driven by a tyrosine hydroxylase promoter in a defective herpes simplex virus amplicon vector. Hum. Gene Ther. 1996, 7, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wang, Y.; Bak, S.Y.; Lang, P.; Ullrey, D.; Neve, R.L.; O'Malley, K.L.; Geller, A.I. An HSV-1 vector containing the rat tyrosine hydroxylase promoter enhances both long-term and cell type-specific expression in the midbrain. J. Neurochem. 1997, 68, 1792–1803. [Google Scholar] [PubMed]
- Rasmussen, M.; Kong, L.; Zhang, G.R.; Liu, M.; Wang, X.; Szabo, G.; Curthoys, N.P.; Geller, A.I. Glutamatergic or GABAergic neuron-specific, long-term expression in neocortical neurons from helper virus-free HSV-1 vectors containing the phosphate-activated glutaminase, vesicular glutamate transporter-1, or glutamic acid decarboxylase promoter. Brain Res. 2007, 1144, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.R.; Wang, X.; Yang, T.; Sun, M.; Zhang, W.; Wang, Y.; Geller, A.I. A tyrosine hydroxylase-neurofilament chimeric promoter enhances long-term expression in rat forebrain neurons from helper virus-free HSV-1 vectors. Brain Res. Mol. Brain Res. 2000, 84, 17–31. [Google Scholar] [CrossRef]
- Gao, Q.; Sun, M.; Wang, X.; Geller, A.I. Isolation of an enhancer from the rat tyrosine hydroxylase promoter that supports long-term, neuronal-specific expression from a neurofilament promoter, in a helper virus-free HSV-1 vector system. Brain Res. 2007, 1130, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Federoff, H.J. Herpes simplex virus type 1 amplicon vectors with glucocorticoid-inducible gene expression. Hum. Gene Ther. 1995, 6, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Bragg, D.C.; Camp, S.M.; Kaufman, C.A.; Wilbur, J.D.; Boston, H.; Schuback, D.E.; Hanson, P.I.; Sena-Esteves, M.; Breakefield, X.O. Perinuclear biogenesis of mutant torsin-A inclusions in cultured cells infected with tetracycline-regulated herpes simplex virus type 1 amplicon vectors. Neuroscience 2004, 125, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Sun, M.; Wang, X.; Zhang, G.R.; Geller, A.I. Long-term inducible expression in striatal neurons from helper virus-free HSV-1 vectors that contain the tetracycline-inducible promoter system. Brain Res. 2006, 1083, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Petravicz, J.; Breakefield, X.O. Single HSV-amplicon vector mediates drug-induced gene expression via dimerizer system. Mol. Ther. 2003, 7, 790–800. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
De Silva, S.; Bowers, W.J. Herpes Virus Amplicon Vectors. Viruses 2009, 1, 594-629. https://doi.org/10.3390/v1030594
De Silva S, Bowers WJ. Herpes Virus Amplicon Vectors. Viruses. 2009; 1(3):594-629. https://doi.org/10.3390/v1030594
Chicago/Turabian StyleDe Silva, Suresh, and William J. Bowers. 2009. "Herpes Virus Amplicon Vectors" Viruses 1, no. 3: 594-629. https://doi.org/10.3390/v1030594
APA StyleDe Silva, S., & Bowers, W. J. (2009). Herpes Virus Amplicon Vectors. Viruses, 1(3), 594-629. https://doi.org/10.3390/v1030594