Murine Coronavirus Cell Type Dependent Interaction with the Type I Interferon Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results and Discussion
2.1. Virus induced innate immune response
2.2. Innate immune response to virus infection in the CNS
2.3. Overview of the interaction of MHV and IFN-α/β pathway
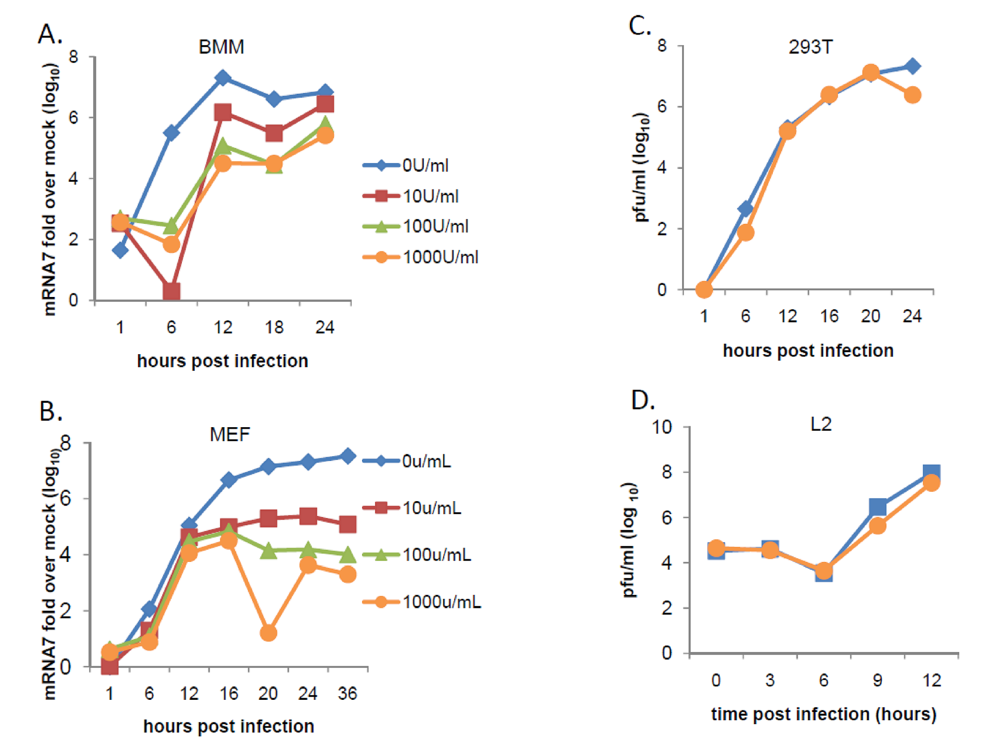
2.4. Sensitivity of MHV to antiviral effects of IFN-α/β is cell-type dependent

2.5. Role of type I IFN signaling during MHV infection in the CNS and liver in vivo
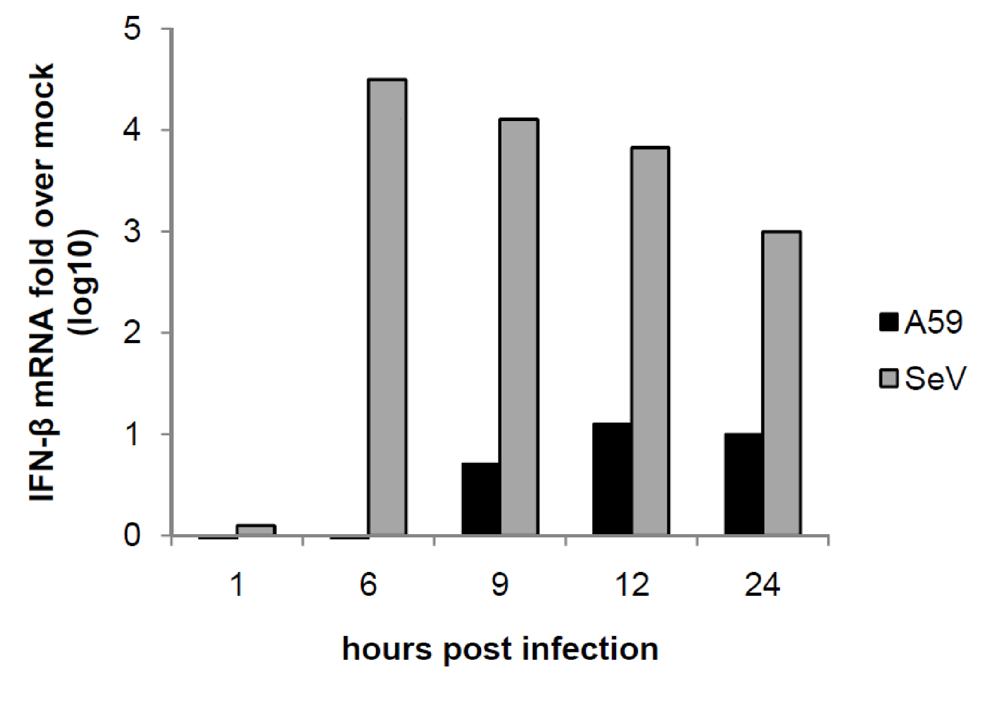
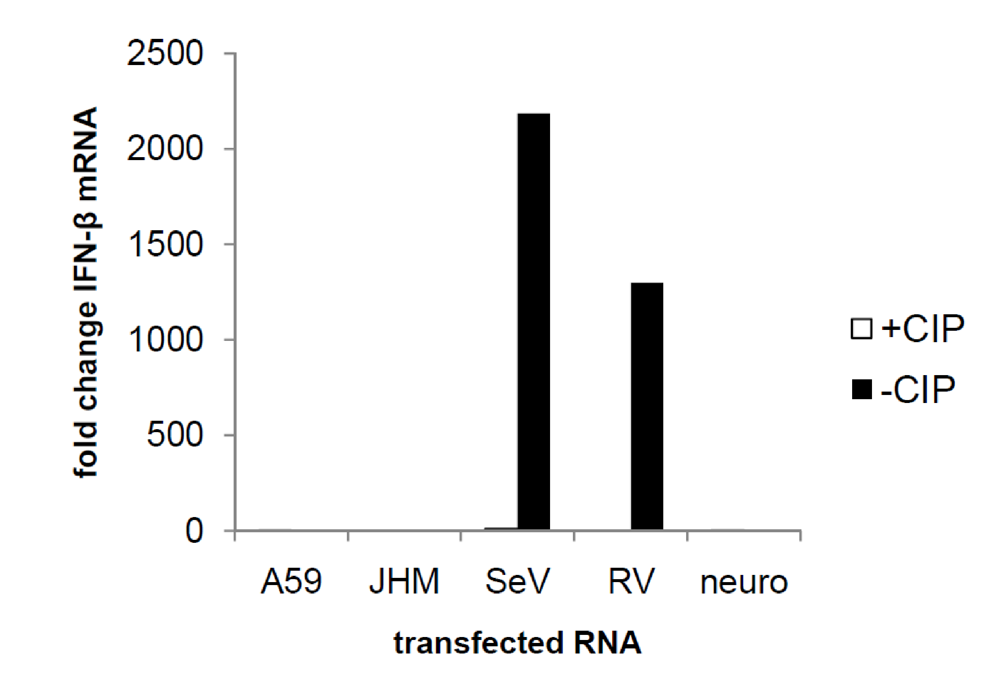
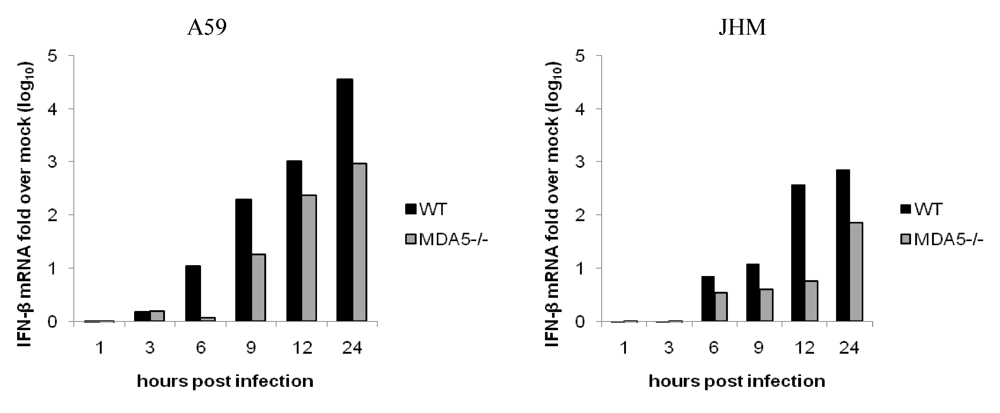
2.6. MHV induction of IFN-α/β



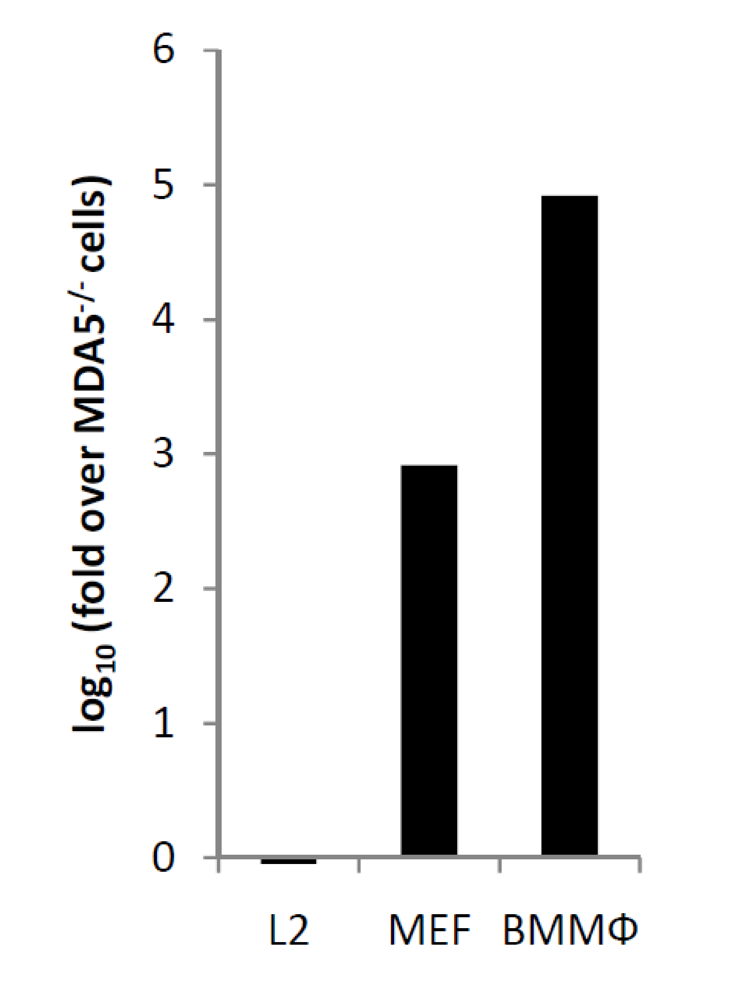
2.7. Induction of IFN-α/β expression in vivo is cell-type specific

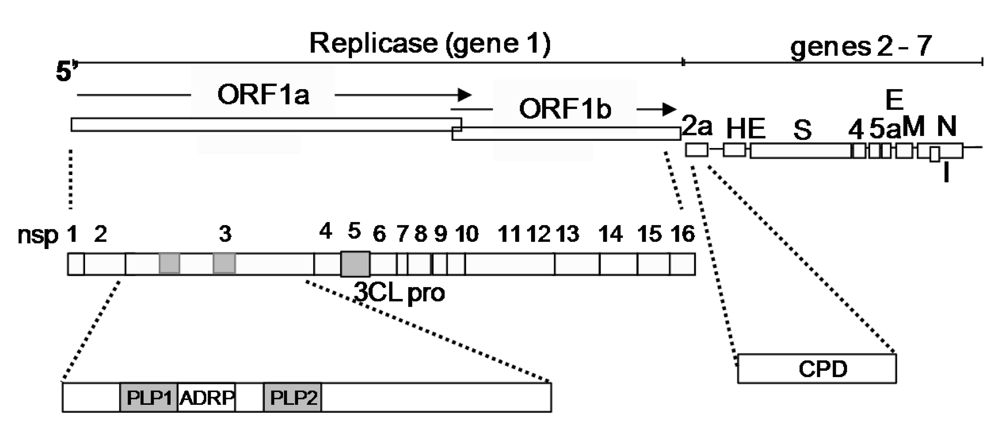
2.8. MHV encoded IFN antagonists
2.9. Nucleocapsid
2.10. Nsp1
2.11. Nsp3
2.12. MHV virulence genes that may play roles other than compromising IFN induction or signaling
2.13. The nsp3 macro domain and ns2 putative cyclophosphodiesterase (CPD)
2.14. Nsp14
3. Conclusions and Future Questions
Acknowledgments
References and Notes
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Phylogenetic and recombination analysis of coronavirus HKU1, a novel coronavirus from patients with pneumonia. Arch. Virol. 2005, 150, 2299–2311. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; Cheng, V.C.; Chan, K.H.; Tsang, D.N.; Yung, R.W.; Ng, T.K.; Yuen, K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Chu, C.M.; Cheng, V.C.; Chan, K.S.; Hung, I.F.; Poon, L.L.; Law, K.I.; Tang, B.S.; Hon, T.Y.; Chan, C.S.; Chan, K.H.; Ng, J.S.; Zheng, B.J.; Ng, W.L.; Lai, R.W.; Guan, Y.; Yuen, K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.S.; Zheng, B.J.; Li, Y.M. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zheng, B.J.; He, Y. Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; Butt, K.M.; Wong, K.L.; Chan, K.W.; Lim, W.; Shortridge, K.F.; Yuen, K.Y.; Peiris, J.S.; Poon, L.L. Isolation and Characterization of Viruses Related to the SARS Coronavirus from Animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Song, H.D.; Tu, C.C.; Zhang, G.W.; Wang, S.Y.; Zheng, K.; Lei, L.C.; Chen, Q.X.; Gao, Y.W.; Zhou, H.Q.; Xiang, H.; Zheng, H.J.; Chern, S.W.; Cheng, F.; Pan, C.M.; Xuan, H.; Chen, S.J.; Luo, H.M.; Zhou, D.H.; Liu, Y.F.; He, J.F.; Qin, P.Z.; Li, L.H.; Ren, Y.Q.; Liang, W.J.; Yu, Y.D.; Anderson, L.; Wang, M.; Xu, R.H.; Wu, X.W.; Zheng, H.Y.; Chen, J.D.; Liang, G.; Gao, Y.; Liao, M.; Fang, L.; Jiang, L.Y.; Li, H.; Chen, F.; Di, B.; He, L.J.; Lin, J.Y.; Tong, S.; Kong, X.; Du, L.; Hao, P.; Tang, H.; Bernini, A.; Yu, X.J.; Spiga, O.; Guo, Z.M.; Pan, H.Y.; He, W.Z.; Manuguerra, J.C.; Fontanet, A.; Danchin, A.; Niccolai, N.; Li, Y.X.; Wu, C.I.; Zhao, G.P. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. U S A 2005, 102, 2430–2435. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Crameri, G.; Kong, X.; Chen, J.; Sun, Y.; Yu, M.; Xiang, H.; Xia, X.; Liu, S.; Ren, T.; Yu, Y.; Eaton, B.T.; Xuan, H.; Wang, L.F. Antibodies to SARS coronavirus in civets. Emerg. Infect. Dis. 2004, 10, 2244–2248. [Google Scholar] [PubMed]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U S A 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; Zhang, J.; McEachern, J.; Field, H.; Daszak, P.; Eaton, B.T.; Zhang, S.; Wang, L.F. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, L.; Sure, K.; Ihorst, G.; Stang, A.; Pyrc, K.; Jebbink, M.F.; Petersen, G.; Forster, J.; Berkhout, B.; Uberla, K. Croup is associated with the novel coronavirus NL63. PLoS Med. 2005, 2, e240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esper, F.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Evidence of a novel human coronavirus that is associated with respiratory tract disease in infants and young children. J. Infect. Dis. 2005, 191, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Homberger, F.R.; Zhang, L.; Barthold, S.W. Prevalence of enterotropic and polytropic mouse hepatitis virus in enzootically infected mouse colonies. Lab. Anim. Sci. 1998, 48, 50–54. [Google Scholar] [PubMed]
- De Albuquerque, N.; Baig, E.; Ma, X.; Zhang, J.; He, W.; Rowe, A.; Habal, M.; Liu, M.; Shalev, I.; Downey, G.P.; Gorczynski, R.; Butany, J.; Leibowitz, J.; Weiss, S.R.; McGilvray, I.D.; Phillips, M.J.; Fish, E.N.; Levy, G.A. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 2006, 80, 10382–10394. [Google Scholar] [CrossRef] [PubMed]
- Spaan, W.; Cavanagh, D.; Horzinek, M.C. Coronaviruses: structure and genome expression. J. Gen. Virol. 1988, 69, 2939–2952. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Peng, D.; Hingley, S.T.; Weiss, S.R.; Masters, P.S. The internal open reading frame within the nucleocapsid gene of mouse hepatitis virus encodes a structural protein that is not essential for viral replication. J. Virol. 1997, 71, 996–1003. [Google Scholar] [PubMed]
- Luytjes, W.; Bredenbeek, P.J.; Noten, A.F.; Horzinek, M.C.; Spaan, W.J. Sequence of mouse hepatitis virus A59 mRNA 2: indications for RNA recombination between coronaviruses and influenza C virus. Virology 1988, 166, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kazi, L.; Lissenberg, A.; Watson, R.; de Groot, R.J.; Weiss, S.R. Expression of hemagglutinin esterase protein from recombinant mouse hepatitis virus enhances neurovirulence. J. Virol. 2005, 79, 15064–15073. [Google Scholar] [CrossRef] [PubMed]
- Marten, N.W.; Stohlman, S.A.; Bergmann, C.C. MHV infection of the CNS: mechanisms of immune-mediated control. Viral Immunol. 2001, 14, 1–18. [Google Scholar] [PubMed]
- Perlman, S.; Dandekar, A.A. Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 2005, 5, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.E.; Weiss, S.R.; Paterson, Y. Murine hepatitis virus--a model for virus-induced CNS demyelination. J. Neurovirol. 2002, 8, 76–85. [Google Scholar] [CrossRef]
- Matsuyama, S.; Henmi, S.; Ichihara, N.; Sone, S.; Kikuchi, T.; Ariga, T.; Taguchi, F. Protective effects of murine recombinant interferon-beta administered by intravenous, intramuscular or subcutaneous route on mouse hepatitis virus infection. Antiviral Res. 2000, 47, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Garlinghouse, L.E.; Smith, A.L.; Holford, T. The biological relationship of mouse hepatitis virus (MHV) strains and interferon: in vitro induction and sensitivities. Arch. Virol. 82, 1984, 19–29. [Google Scholar]
- Stohlman, S.A.; Sakaguchi, A.Y.; Hiti, A. Interferon production and activity in mouse neuroblastoma cells. Arch. Virol. 1978, 57, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Haluskey, J.A.; Lavi, E. Coronavirus MHV-A59 causes upregulation of interferon-beta RNA in primary glial cell cultures. Adv. Exp. Med. Biol. 1998, 440, 451–454. [Google Scholar] [PubMed]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Huang, C.; Makino, S. SARS coronavirus accessory proteins. Virus Res. 2008, 133, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Baric, R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol. Mol. Biol. Rev. 2008, 72 (Table of Contents.), 672–685. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Heise, M.; Baric, R. SARS coronavirus and innate immunity. Virus Res. 2008, 133, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Weber, F. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev. 2008, 19, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Ricour, C.; Sommereyns, C.; Sorgeloos, F.; Michiels, T. Type I interferon response in the central nervous system. Biochimie 2007, 89, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; Bredenbeek, P.J.; van den Worm, S.H.; Spaan, W.J. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology 2007, 361, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Hingley, S.T.; Simmons, G.; Yu, C.; Das Sarma, J.; Bates, P.; Weiss, S.R. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 2006, 80, 5768–5776. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Lee, J.O. Structures of the toll-like receptor family and its ligand complexes. Immunity 2008, 29, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Gale, Jr., M. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J. Exp. Med. 2008, 205, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition in the innate immune response. Biochem. J. 2009, 420, 1–16. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Delhaye, S.; Paul, S.; Blakqori, G.; Minet, M.; Weber, F.; Staeheli, P.; Michiels, T. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U S A 2006, 103, 7835–7840. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Perlman, S. Mouse hepatitis virus does not induce Beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 2007, 81, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Roth-Cross, J.K.; Martinez-Sobrido, L.; Scott, E.P.; Garcia-Sastre, A.; Weiss, S.R. Inhibition of the IFN-a/b Response by Mouse Hepatitis Virus (MHV) at Multiple Levels. J. Virol. 2007, 81, 7189–7199. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Hauns, K.; Langland, J.O.; Jacobs, B.L.; Hogue, B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007, 81, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Ireland, D.D.; Stohlman, S.A.; Hinton, D.R.; Atkinson, R.; Bergmann, C.C. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 2008, 82, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Zust, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Kalinke, U.; Zust, R.; Konig, M.; Reizis, B.; Lopez-Macias, C.; Thiel, V.; Ludewig, B. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J. Immunol. 2009, 182, 1099–1106. [Google Scholar] [PubMed]
- Roth-Cross, J.K.; Bender, S.J.; Weiss, S.R. Murine Coronavirus Mouse Hepatitis Virus (MHV) is Recognized by MDA5 and Induces Type I IFN in Brain Macrophages/Microglia. J. Virol. 2008. [Google Scholar]
- Virelizier, J.L.; Gresser, I. Role of interferon in the pathogenesis of viral diseases of mice as demonstrated by the use of anti-interferon serum. V. Protective role in mouse hepatitis virus type 3 infection of susceptible and resistant strains of mice. J. Immunol. 1978, 120, 1616–1619. [Google Scholar]
- Fuchizaki, U.; Kaneko, S.; Nakamoto, Y.; Sugiyama, Y.; Imagawa, K.; Kikuchi, M.; Kobayashi, K. Synergistic antiviral effect of a combination of mouse interferon-alpha and interferon-gamma on mouse hepatitis virus. J. Med. Virol. 2003, 69, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Noda, Y.; Unoura, M.; Tanaka, N.; Kobayashi, K.; Hattori, N.; Hatano, K.; Kobayashi, S. Effect of exogenous mouse interferon on murine fulminant hepatitis induced by mouse hepatitis virus type 2. Dig. Dis. Sci. 1986, 31, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Sone, S.; Izawa, A.; Narumi, H.; Kajita, A.; Tanabe, J.; Taguchi, F. The combination of type I interferon and ribavirin has an inhibitory effect on mouse hepatitis virus infection. Hepatol. Res. 2007, 37, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Aurisicchio, L.; Delmastro, P.; Salucci, V.; Paz, O.G.; Rovere, P.; Ciliberto, G.; La Monica, N.; Palombo, F. Liver-specific alpha 2 interferon gene expression results in protection from induced hepatitis. J. Virol. 2000, 74, 4816–4823. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Sinclair, S.B.; Fung, L.S.; Cole, E.H.; Levy, G.A. Effect of eicosanoids on induction of procoagulant activity by murine hepatitis virus strain 3 in vitro. Prostaglandins 1991, 42, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Zust, R.; Cervantes-Barragan, L.; Kuri, T.; Blakqori, G.; Weber, F.; Ludewig, B.; Thiel, V. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007, 3, e109. [Google Scholar] [CrossRef] [PubMed]
- Zurney, J.; Howard, K.E.; Sherry, B. Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac antiviral responses. J. Virol. 2007, 81, 13668–13680. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.E.; Daley, J.K.; Rall, G.F. Neuronal survival strategies in the face of RNA viral infection. J. Infect. Dis. 2002, 186, S215–S219. [Google Scholar] [CrossRef] [PubMed]
- Macnamara, K.C.; Bender, S.J.; Chua, M.M.; Watson, R.; Weiss, S.R. Priming of CD8+ T cells during central nervous system infection with a murine coronavirus is strain-dependent. J. Virol. 2008, 6150–6160. [Google Scholar] [CrossRef]
- Rempel, J.D.; Murray, S.J.; Meisner, J.; Buchmeier, M.J. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology 2004, 318, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Iacono, K.T.; Kazi, L.; Weiss, S.R. Both spike and background genes contribute to murine coronavirus neurovirulence J. Virol. 2006, 80, 6834–6843. [Google Scholar] [CrossRef]
- Glass, W.G.; Chen, B.P.; Liu, M.T.; Lane, T.E. Mouse hepatitis virus infection of the central nervous system: chemokine-mediated regulation of host defense and disease. Viral Immunol. 2002, 15, 261–272. [Google Scholar] [PubMed]
- Detje, C.N.; Meyer, T.; Schmidt, H.; Kreuz, D.; Rose, J.K.; Bechmann, I.; Prinz, M.; Kalinke, U. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 2009, 182, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Pichlmair, A.; Martinez-Sobrido, L.; Cros, J.; Garcia-Sastre, A.; Haller, O.; Weber, F. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 2005, 79, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Barrett, J.W.; Ma, Y.; Dekaban, G.A.; McFadden, G. Induction of alpha/beta interferon by myxoma virus is selectively abrogated when primary mouse embryo fibroblasts become immortalized. J. Virol. 2009, 83, 5928–5932. [Google Scholar] [CrossRef] [PubMed]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef] [PubMed]
- Brockway, S.M.; Lu, X.T.; Peters, T.R.; Dermody, T.S.; Denison, M.R. Intracellular localization and protein interactions of the gene 1 protein p28 during mouse hepatitis virus replication. J. Virol. 2004, 78, 11551–11562. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Keller, B.C.; Gale, M.; Diamond, M.S. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J. Virol. 2008, 82, 8465–8475. [Google Scholar] [CrossRef] [PubMed]
- Flanegan, J.B.; Petterson, R.F.; Ambros, V.; Hewlett, N.J.; Baltimore, D. Covalent linkage of a protein to a defined nucleotide sequence at the 5'-terminus of virion and replicative intermediate RNAs of poliovirus. Proc. Natl. Acad. Sci. U S A 1977, 74, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; Mirazimi, A.; Weber, F. Processing of genome 5' termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Bamming, D.; Horvath, C.M. Negative regulation of cytoplasmic RNA-mediated antiviral signaling. Cytokine 2008, 43, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. RIG-I is cleaved during picornavirus infection. Virology 2009, 39, 171–6. [Google Scholar] [CrossRef]
- Rempel, J.D.; Murray, S.J.; Meisner, J.; Buchmeier, M.J. Mouse hepatitis virus neurovirulence: evidence of a linkage between S glycoprotein expression and immunopathology. Virology 2004, 318, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Huang, C.; Lokugamage, K.; Kamitani, W.; Ikegami, T.; Tseng, C.T.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008, 82, 4471–4479. [Google Scholar] [CrossRef] [PubMed]
- Wathelet, M.G.; Orr, M.; Frieman, M.B.; Baric, R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007, 81, 11620–11633. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; Li, K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Chen, G.; Guo, B.; Cheng, G.; Tang, H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008, 18, 1105–1113. [Google Scholar] [PubMed]
- Changolkar, L.N.; Singh, G.; Pehrson, J.R. macroH2A1-dependent silencing of endogenous murine leukemia viruses. Mol. Cell Biol. 2008, 28, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Koonin, E.V.; Lai, M.M. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 1991, 288, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.; Guan, Y.; Rozanov, M.; Spaan, W.J.; Gorbalenya, A.E. Unique and Conserved Features of Genome and Proteome of SARS-coronavirus, an Early Split-off From the Coronavirus Group 2 Lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Putics, A.; Filipowicz, W.; Hall, J.; Gorbalenya, A.E.; Ziebuhr, J. ADP-ribose-1"-monophosphatase: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J. Virol. 2005, 79, 12721–12731. [Google Scholar] [CrossRef] [PubMed]
- Putics, A.; Gorbalenya, A.E.; Ziebuhr, J. Identification of protease and ADP-ribose 1''-monophosphatase activities associated with transmissible gastroenteritis virus non-structural protein 3. J. Gen. Virol. 2006, 87, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.P.; Malet, H.; Putics, A.; Heinonen, M.; Dutartre, H.; Frangeul, A.; Gruez, A.; Campanacci, V.; Cambillau, C.; Ziebuhr, J.; Ahola, T.; Canard, B. Structural and Functional Basis for ADP-Ribose and Poly(ADP-Ribose) Binding by Viral Macro Domains. J. Virol. 2006, 80, 8493–8502. [Google Scholar] [CrossRef] [PubMed]
- Roth-Cross, J.K.; Stokes, H.; Chang, G.; Chua, M.M.; Thiel, V.; Weiss, S.R.; Gorbalenya, A.E.; Siddell, S.G. Organ-specific attenuation of murine hepatitis virus strain A59 by replacement of catalytic residues in the putative viral cyclic phosphodiesterase ns2. J. Virol. 2009, 83, 3743–3753. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.K.; Cervantes-Barragan, L.; Ludewig, B.; Thiel, V. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1''-phosphatase, a viral function conserved in the alpha-like supergroup. J. Virol. 2008, 82, 12325–12334. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Smith, F.D.; Scott, J.D.; Barford, D. AKAP18 contains a phosphoesterase domain that binds AMP. J. Mol. Biol. 2008, 375, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, Y.; Hansen, G.; Boomaars-van der Zanden, A.L.; Snijder, E.J.; Gorbalenya, A.E.; Hilgenfeld, R. Crystal structures of the X-domains of a Group-1 and a Group-3 coronavirus reveal that ADP-ribose-binding may not be a conserved property. Protein Sci. 2009, 18, 6–16. [Google Scholar] [PubMed]
- Park, E.; Griffin, D.E. Interaction of Sindbis virus non-structural protein 3 with poly(ADP-ribose) polymerase 1 in neuronal cells. J. Gen. Virol. 2009, 90, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Griffin, D.E. The nsP3 macro domain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 2009, 388, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef] [PubMed]
- Sperry, S.; Kazi, L.; Graham, R.; Baric, R.; Weiss, S.; Denison, M. Single amino acid substitutions in non-structural ORF1b-nsp14 and ORF2a 30kDa proteins of the murine coronavirus MHV-A59 are attenuating in mice. J. Virol. 2005, 79, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Rose, K.M.; Weiss, S.R. Murine Coronavirus Cell Type Dependent Interaction with the Type I Interferon Response. Viruses 2009, 1, 689-712. https://doi.org/10.3390/v1030689
Rose KM, Weiss SR. Murine Coronavirus Cell Type Dependent Interaction with the Type I Interferon Response. Viruses. 2009; 1(3):689-712. https://doi.org/10.3390/v1030689
Chicago/Turabian StyleRose, Kristine M., and Susan R. Weiss. 2009. "Murine Coronavirus Cell Type Dependent Interaction with the Type I Interferon Response" Viruses 1, no. 3: 689-712. https://doi.org/10.3390/v1030689
APA StyleRose, K. M., & Weiss, S. R. (2009). Murine Coronavirus Cell Type Dependent Interaction with the Type I Interferon Response. Viruses, 1(3), 689-712. https://doi.org/10.3390/v1030689