Distinct Effects of Type I and III Interferons on Enteric Viruses



Abstract
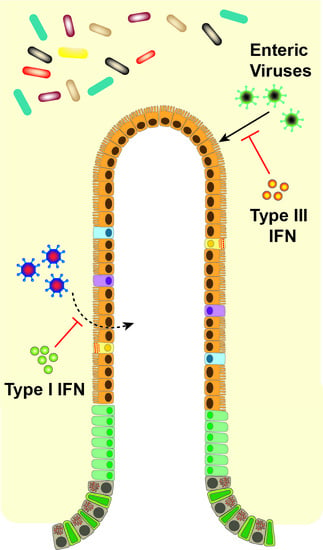
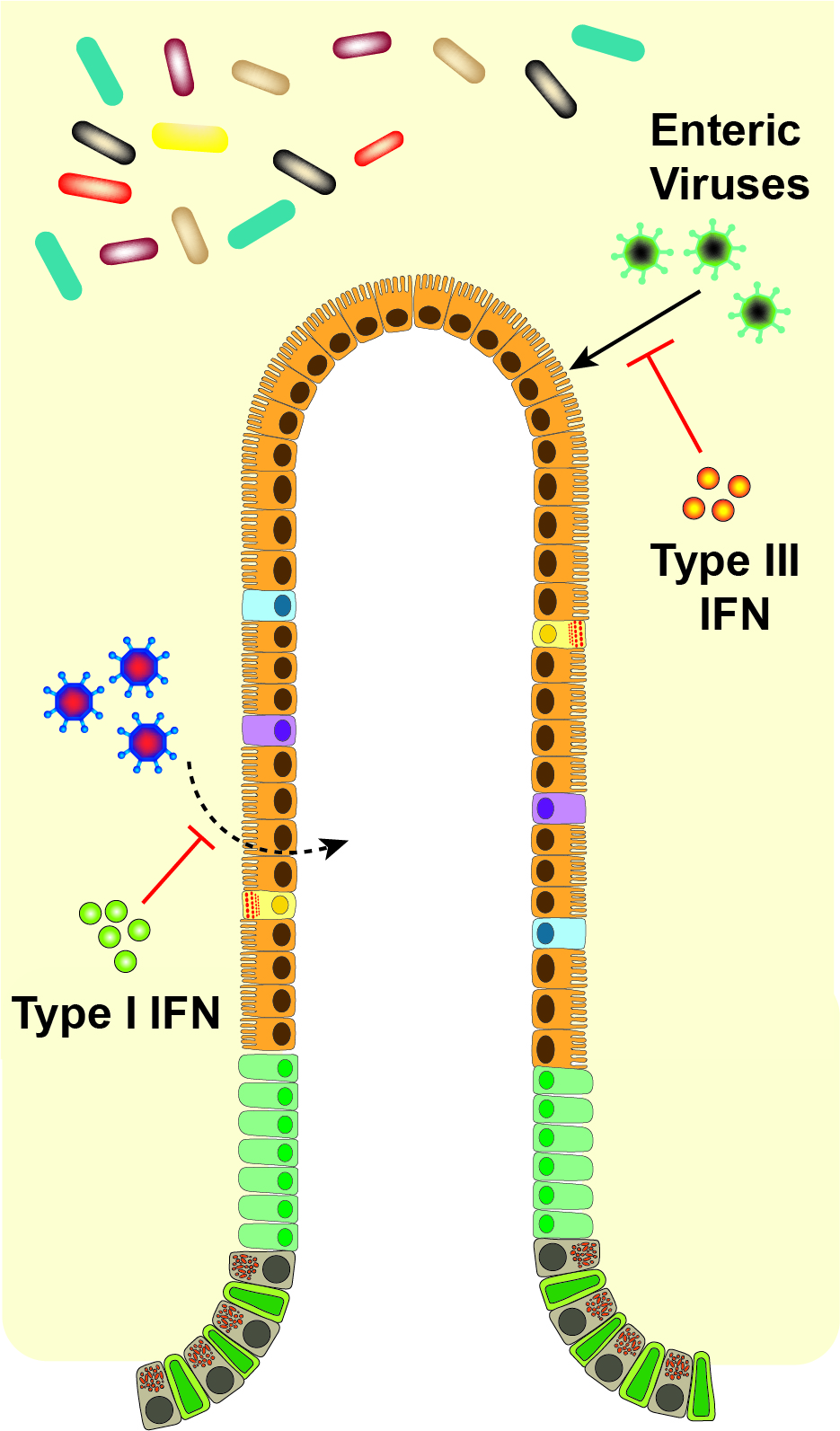
:
1. Introduction
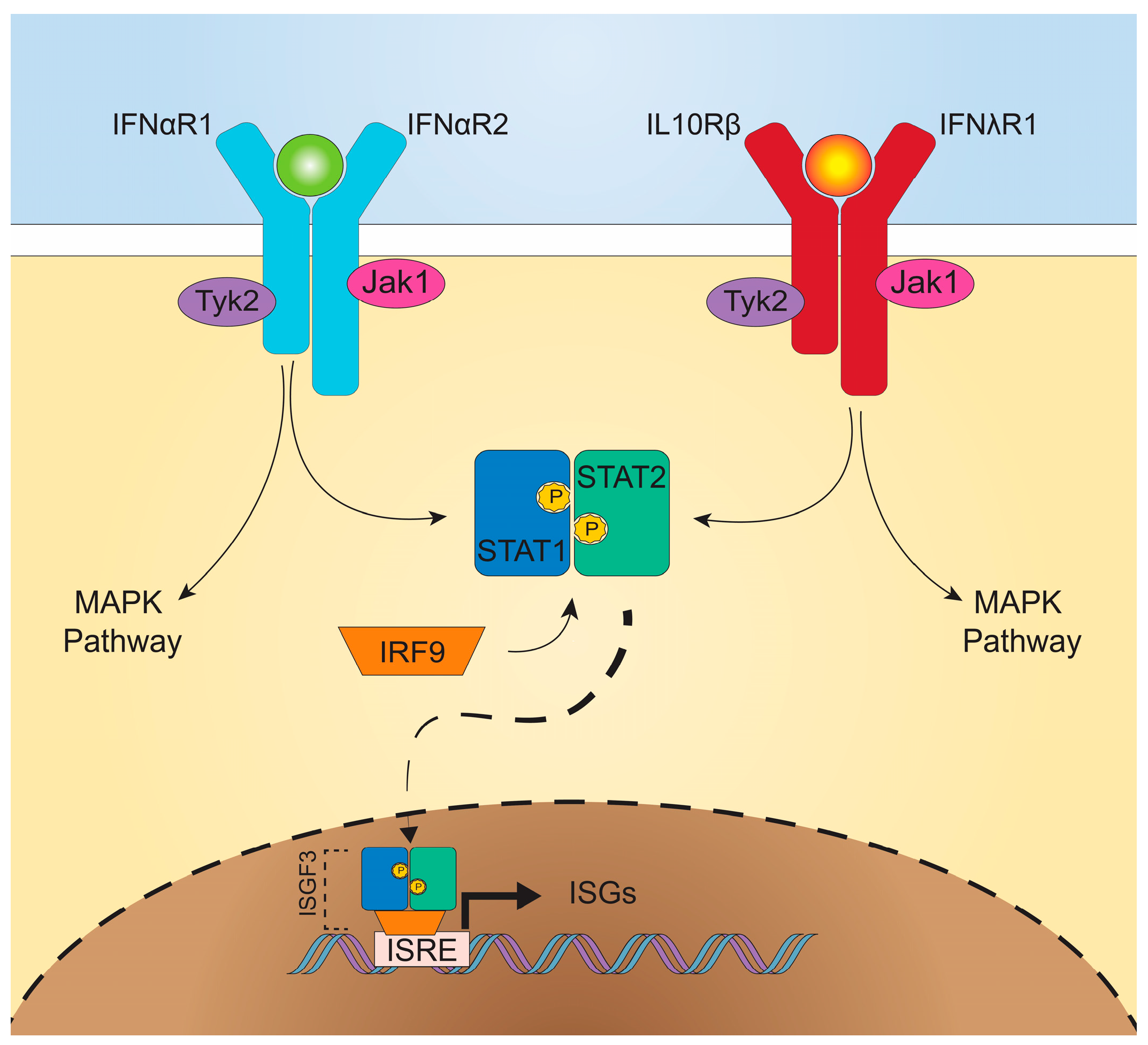
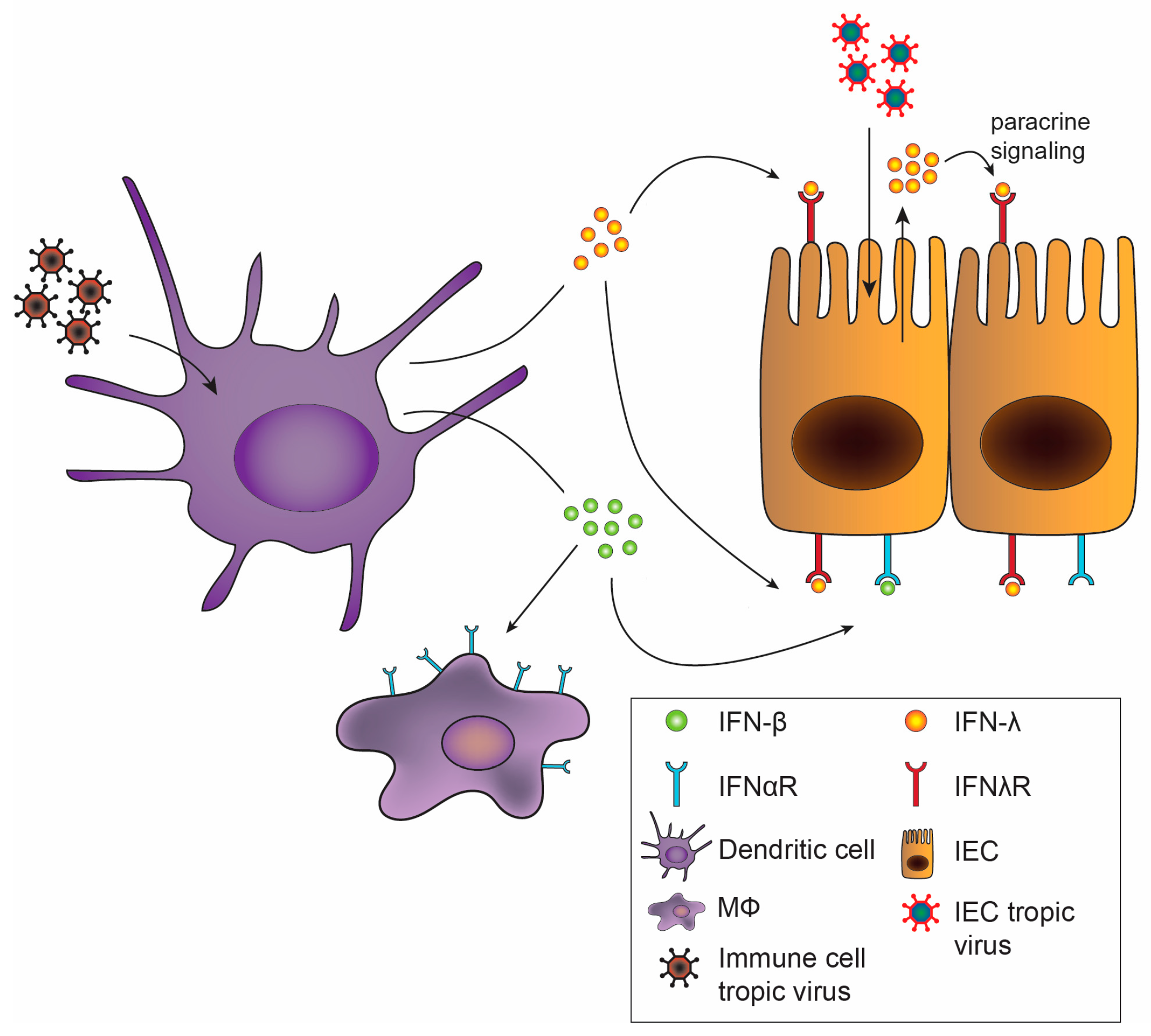
2. Type I Versus Type III Interferons
3. Differential Control of Individual Viruses by Type I and III IFNs
4. Individual Interferon Stimulated Genes
5. Viral Evasion and Antagonism Strategies
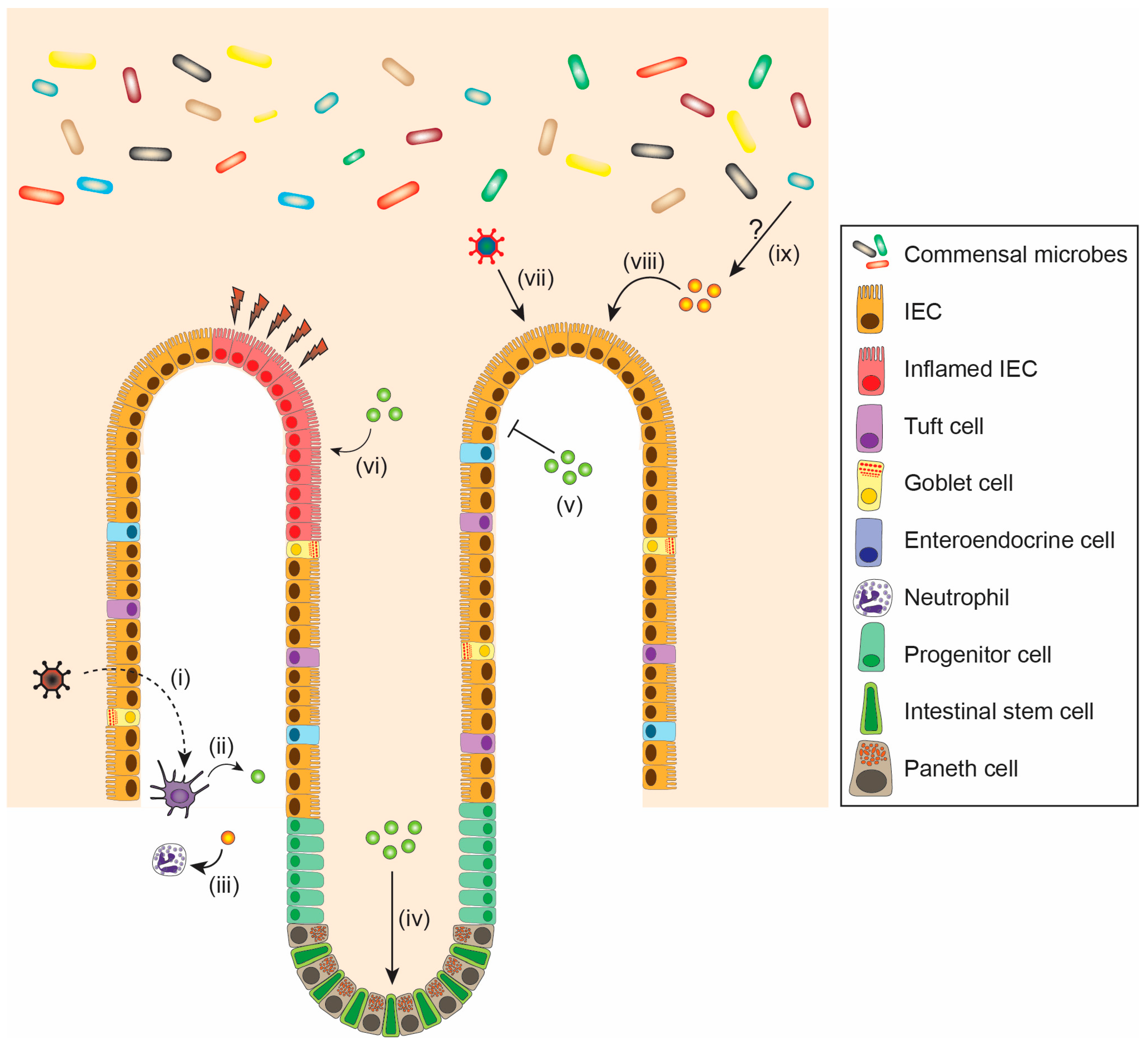
6. Physiological Effects of IFNs on the Intestine
7. IFNs and the Microbiota
8. Type I and III IFNs at other Barriers
9. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Alsharifi, M.; Mullbacher, A.; Regner, M. Interferon type I responses in primary and secondary infections. Immunol. Cell Biol. 2008, 86, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Santer, D.M.; O’Shea, D.; Tyrrell, D.L.; Houghton, M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg. Microbes Infect. 2014, 3, e51. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.G. Structure and function of the gut. In Oxford Textbook of Medicine; Warrell, D.A., Cox, T.M., Firth, J.D., Eds.; Oxford University Press: Oxford, UK, 2010; pp. 2201–2204. [Google Scholar]
- Lengyel, P. Biochemistry of interferons and their actions. Annu. Rev. Biochem. 1982, 51, 251–282. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- De Weerd, N.A.; Vivian, J.P.; Nguyen, T.K.; Mangan, N.E.; Gould, J.A.; Braniff, S.J.; Zaker-Tabrizi, L.; Fung, K.Y.; Forster, S.C.; Beddoe, T.; et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor ifnar1. Nat. Immunol. 2013, 14, 901–907. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Roisman, L.C.; Jaks, E.; Gavutis, M.; Piehler, J.; van der Heyden, J.; Uze, G.; Schreiber, G. Inquiring into the differential action of interferons (IFNs): An IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol. Cell. Biol. 2006, 26, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- De Weerd, N.A.; Matthews, A.Y.; Pattie, P.R.; Bourke, N.M.; Lim, S.S.; Vivian, J.P.; Rossjohn, J.; Hertzog, P.J. A hot spot on interferon alpha/beta receptor subunit 1 (IFNAR1) underpins its interaction with interferon-beta and dictates signaling. J. Biol. Chem. 2017, 292, 7554–7565. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class ii cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Hemann, E.A.; Gale, M., Jr.; Savan, R. Interferon lambda genetics and biology in regulation of viral control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Baldridge, M.T. Interferon-lambda: A potent regulator of intestinal viral infections. Front. Immunol. 2017, 8, 749. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Dumoutier, L.; Tounsi, A.; Michiels, T.; Sommereyns, C.; Kotenko, S.V.; Renauld, J.C. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: Similarities with type i interferon signaling. J. Biol. Chem. 2004, 279, 32269–32274. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAKSTAT 2013, 2, e23931. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the JAK-STAT pathway and the mitogen-activated protein kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.D.; Feng, N.; Sen, A.; Balan, M.; Tseng, H.C.; McElrath, C.; Smirnov, S.V.; Peng, J.; Yasukawa, L.L.; Durbin, R.K.; et al. Distinct roles of type I and type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog. 2016, 12, e1005600. [Google Scholar]
- Selvakumar, T.A.; Bhushal, S.; Kalinke, U.; Wirth, D.; Hauser, H.; Koster, M.; Hornef, M.W. Identification of a predominantly interferon-lambda-induced transcriptional profile in murine intestinal epithelial cells. Front. Immunol. 2017, 8, 1302. [Google Scholar] [CrossRef] [PubMed]
- Novatt, H.; Theisen, T.C.; Massie, T.; Massie, T.; Simonyan, V.; Voskanian-Kordi, A.; Renn, L.A.; Rabin, R.L. Distinct patterns of expression of transcription factors in response to interferonbeta and interferonlambda1. J. Interferon Cytokine Res. 2016, 36, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Voigt, E.A.; Yin, J. Kinetic differences and synergistic antiviral effects between type I and type III interferon signaling indicate pathway independence. J. Interferon Cytokine Res. 2015, 35, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Bolen, C.R.; Ding, S.; Robek, M.D.; Kleinstein, S.H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 2014, 59, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Jilg, N.; Lin, W.; Hong, J.; Schaefer, E.A.; Wolski, D.; Meixong, J.; Goto, K.; Brisac, C.; Chusri, P.; Fusco, D.N.; et al. Kinetic differences in the induction of interferon stimulated genes by interferon-alpha and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 2014, 59, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, S.; Guan, Y.; Chen, L. Type III interferon induces distinct SOCS1 expression pattern that contributes to delayed but prolonged activation of JAK/STAT signaling pathway: Implications for treatment non-response in hcv patients. PLoS ONE 2015, 10, e0133800. [Google Scholar] [CrossRef] [PubMed]
- Blumer, T.; Coto-Llerena, M.; Duong, F.H.T.; Heim, M.H. SOCS1 is an inducible negative regulator of interferon lambda (IFN-lambda)-induced gene expression in vivo. J. Biol. Chem. 2017, 292, 17928–17938. [Google Scholar] [CrossRef] [PubMed]
- Pervolaraki, K.; Stanifer, M.L.; Munchau, S.; Renn, L.A.; Albrecht, D.; Kurzhals, S.; Senis, E.; Grimm, D.; Schroder-Braunstein, J.; Rabin, R.L.; et al. Type I and type III interferons display different dependency on mitogen-activated protein kinases to mount an antiviral state in the human gut. Front. Immunol. 2017, 8, 459. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Severa, M.; Giacomini, E.; Monneron, D.; Remoli, M.E.; Julkunen, I.; Cella, M.; Lande, R.; Uze, G. Viral infection and toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004, 34, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Mahlakoiv, T.; Hernandez, P.; Gronke, K.; Diefenbach, A.; Staeheli, P. Leukocyte-derived IFN-alpha/beta and epithelial IFN-lambda constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015, 11, e1004782. [Google Scholar] [CrossRef] [PubMed]
- Jewell, N.A.; Cline, T.; Mertz, S.E.; Smirnov, S.V.; Flano, E.; Schindler, C.; Grieves, J.L.; Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Lambda interferon is the predominant interferon induced by influenza a virus infection in vivo. J. Virol. 2010, 84, 11515–11522. [Google Scholar] [CrossRef] [PubMed]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Montoya, M.; Unger, H.; Alexopoulou, L.; Roy, P.; Haswell, L.E.; Al-Shamkhani, A.; Flavell, R.; Borrow, P.; Reis e Sousa, C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003, 424, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.; Juleff, N.; Windsor, M.; Gubbins, S.; Roberts, L.; Morgan, S.; Meyers, G.; Perez-Martin, E.; Tchilian, E.; Charleston, B.; et al. Type I and III IFNs produced by plasmacytoid dendritic cells in response to a member of the flaviviridae suppress cellular immune responses. J. Immunol. 2016, 196, 4214–4226. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Durbin, J.E. Contribution of type III interferons to antiviral immunity: Location, location, location. J. Biol. Chem. 2017, 292, 7295–7303. [Google Scholar] [CrossRef] [PubMed]
- Pulverer, J.E.; Rand, U.; Lienenklaus, S.; Kugel, D.; Zietara, N.; Kochs, G.; Naumann, R.; Weiss, S.; Staeheli, P.; Hauser, H.; et al. Temporal and spatial resolution of type I and III interferon responses in vivo. J. Virol. 2010, 84, 8626–8638. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; Lee, S.; Brown, J.J.; McAllister, N.; Urbanek, K.; Dermody, T.S.; Nice, T.J.; Virgin, H.W. Expression of IFNLR1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against norovirus and reovirus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Bhushal, S.; Wolfsmuller, M.; Selvakumar, T.A.; Kemper, L.; Wirth, D.; Hornef, M.W.; Hauser, H.; Koster, M. Cell polarization and epigenetic status shape the heterogeneous response to type III interferons in intestinal epithelial cells. Front. Immunol. 2017, 8, 671. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Wetzel, J.D.; He, J.; Mikacenic, C.; Dermody, T.S.; Kelsall, B.L. Type I interferons produced by hematopoietic cells protect mice against lethal infection by mammalian reovirus. J. Exp. Med. 2007, 204, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Goel-Apaza, S.; Espetia, S.; Velasquez, D.; Cabrera, L.; Loli, S.; Crabtree, J.E.; Black, R.E.; Kosek, M.; Checkley, W.; et al. Multiple norovirus infections in a birth cohort in a peruvian periurban community. Clin. Infect. Dis. 2014, 58, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; Turula, H.; Wobus, C.E. Norovirus regulation by host and microbe. Trends Mol. Med. 2016, 22, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Nice, T.J.; Strong, D.W.; McCune, B.T.; Pohl, C.S.; Virgin, H.W. A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J. Virol. 2013, 87, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Karst, S.M.; Wobus, C.E.; Lay, M.; Davidson, J.; Virgin, H.W. STAT1-dependent innate immunity to a norwalk-like virus. Science 2003, 299, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Thackray, L.B.; Duan, E.; Lazear, H.M.; Kambal, A.; Schreiber, R.D.; Diamond, M.S.; Virgin, H.W. Critical role for interferon regulatory factor 3 (IRF-3) and IRF-7 in type I interferon-mediated control of murine norovirus replication. J. Virol. 2012, 86, 13515–13523. [Google Scholar] [CrossRef] [PubMed]
- Nice, T.J.; Osborne, L.C.; Tomov, V.T.; Artis, D.; Wherry, E.J.; Virgin, H.W. Type I interferon receptor deficiency in dendritic cells facilitates systemic murine norovirus persistence despite enhanced adaptive immunity. PLoS Pathog. 2016, 12, e1005684. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Regev, D.; Watanabe, M.; Hickman, D.; Moussatche, N.; Jesus, D.M.; Kahan, S.M.; Napthine, S.; Brierley, I.; Hunter, R.N., III; et al. Identification of immune and viral correlates of norovirus protective immunity through comparative study of INTRA-cluster norovirus strains. PLoS Pathog. 2013, 9, e1003592. [Google Scholar] [CrossRef] [PubMed]
- Wobus, C.E.; Karst, S.M.; Thackray, L.B.; Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Krug, A.; Mackenzie, J.M.; Green, K.Y.; Virgin, H.W. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004, 2, e432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau, K.R.; Roth, A.N.; Zhu, S.; Hernandez, A.; Colliou, N.; DiVita, B.B.; Philip, D.T.; Riffe, C.; Giasson, B.; Wallet, S.M.; et al. The major targets of acute norovirus infection are immune cells in the gut-associated lymphoid tissue. Nat. Microbiol. 2017, 2, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Wilen, C.B.; Orvedahl, A.; McCune, B.T.; Kim, K.W.; Orchard, R.C.; Peterson, S.T.; Nice, T.J.; Baldridge, M.T.; Virgin, H.W. Norovirus cell tropism is determined by combinatorial action of a viral non-structural protein and host cytokine. Cell Host Microbe 2017, 22, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Robertson, S.J. IFN-lambda: The key to norovirus’s secret hideaway. Cell Host Microbe 2017, 22, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Tomov, V.T.; Palko, O.; Lau, C.W.; Pattekar, A.; Sun, Y.; Tacheva, R.; Bengsch, B.; Manne, S.; Cosma, G.L.; Eisenlohr, L.C.; et al. Differentiation and protective capacity of virus-specific CD8+ T cells suggest murine norovirus persistence in an immune-privileged enteric niche. Immunity 2017, 47, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 2015, 347, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Pereira, J.; Jacobs, S.; Noppen, S.; Verbeken, E.; Michiels, T.; Neyts, J. Interferon lambda (IFN-lambda) efficiently blocks norovirus transmission in a mouse model. Antivir. Res. 2017, 149, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Hiller, B.E.; Thete, D.; Snyder, A.J.; Perez, E., Jr.; Upton, J.W.; Danthi, P. Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Angel, J.; Franco, M.A.; Greenberg, H.B.; Bass, D. Lack of a role for type I and type II interferons in the resolution of rotavirus-induced diarrhea and infection in mice. J. Interferon Cytokine Res. 1999, 19, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.P.; Mahlakoiv, T.; Yang, I.; Schwierzeck, V.; Nguyen, N.; Guendel, F.; Gronke, K.; Ryffel, B.; Hoelscher, C.; Dumoutier, L.; et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 2015, 16, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Simon, L.M.; Zeng, X.L.; Blutt, S.E.; Crawford, S.E.; Sastri, N.P.; Karandikar, U.C.; Ajami, N.J.; Zachos, N.C.; Kovbasnjuk, O.; et al. A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc. Natl. Acad. Sci. USA 2017, 114, E570–E579. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Kim, B.; Fenaux, M.; Nguyen, H.; Vo, P.; Omary, M.B.; Greenberg, H.B. Role of interferon in homologous and heterologous rotavirus infection in the intestines and extraintestinal organs of suckling mice. J. Virol. 2008, 82, 7578–7590. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus detection by the CGAS/STING/TBK1 DNA sensing cascade. J. Virol. 2014, 88, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; Burgert, H.G.; Chen, Y.H.; Sanghera, S.; Katafigiotis, S.; Randall, R.E.; Connerton, I.; Mellits, K.H. Improved growth of enteric adenovirus type 40 in a modified cell line that can no longer respond to interferon stimulation. J. Gen. Virol. 2007, 88, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, V.; King, E.; Totemeyer, S.; Connerton, I.; Mellits, K.H. Interferon treatment suppresses enteric adenovirus infection in a model gastrointestinal cell-culture system. J. Gen. Virol. 2012, 93, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, C.; Eisenacher, K.; Gross, O.; Reindl, W.; Schmitz, F.; Ruland, J.; Krug, A. The IFN regulatory factor 7-dependent type I IFN response is not essential for early resistance against murine cytomegalovirus infection. Eur. J. Immunol. 2009, 39, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Beigel, F.; Olszak, T.; Zitzmann, K.; Eichhorst, S.T.; Otte, J.M.; Diebold, J.; Diepolder, H.; Adler, B.; Auernhammer, C.J.; et al. IL-28a and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G960–G968. [Google Scholar] [CrossRef] [PubMed]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Tissera, M.S.; Cowley, D.; Bogdanovic-Sakran, N.; Hutton, M.L.; Lyras, D.; Kirkwood, C.D.; Buttery, J.P. Options for improving effectiveness of rotavirus vaccines in developing countries. Hum. Vaccines Immunother. 2017, 13, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Rothenberg, M.E.; Mukherjee, G.; Feng, N.; Kalisky, T.; Nair, N.; Johnstone, I.M.; Clarke, M.F.; Greenberg, H.B. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proc. Natl. Acad. Sci. USA 2012, 109, 20667–20672. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Calle, I.; Gomez-Rial, J.; Martinon-Torres, F. Systemic features of rotavirus infection. J. Infect. 2016, 72, S98–S105. [Google Scholar] [CrossRef] [PubMed]
- Echavarria, M. Adenoviruses in immunocompromised hosts. Clin. Microbiol. Rev. 2008, 21, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.J.; Hallett, R.; Tullo, A.B.; Klapper, P.E. The epidemiology of adenovirus infections in greater manchester, UK 1982-96. Epidemiol. Infect. 2000, 125, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.P.; Brandt, C.D.; Wassermann, F.E.; Hall, C.E.; Spigland, I.; Kogon, A.; Elveback, L.R. The virus watch program: A continuing surveillance of viral infections in metropolitan new york families. VI. Observations of adenovirus infections: Virus excretion patterns, antibody response, efficiency of surveillance, patterns of infections, and relation to illness. Am. J. Epidemiol. 1969, 89, 25–50. [Google Scholar] [PubMed]
- Kosulin, K.; Geiger, E.; Vecsei, A.; Huber, W.D.; Rauch, M.; Brenner, E.; Wrba, F.; Hammer, K.; Innerhofer, A.; Potschger, U.; et al. Persistence and reactivation of human adenoviruses in the gastrointestinal tract. Clin. Microbiol. Infect. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Calcedo, R.; Medina-Jaszek, A.; Keough, M.; Peng, H.; Wilson, J.M. Adenoviruses in lymphocytes of the human gastro-intestinal tract. PLoS ONE 2011, 6, e24859. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Razonable, R.R. Cytomegalovirus infections in solid organ transplantation: A review. Infect. Chemother. 2013, 45, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Miyoshi, H.; Origanti, S.; Nice, T.J.; Barger, A.C.; Manieri, N.A.; Fogel, L.A.; French, A.R.; Piwnica-Worms, D.; Piwnica-Worms, H.; et al. Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe 2015, 17, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Sorgeloos, F.; Caddy, S.L.; Vashist, S.; Sosnovtsev, S.; Lloyd, R.; Heesom, K.; Locker, N.; Goodfellow, I. Norovirus-mediated modification of the translational landscape via virus and host-induced cleavage of translation initiation factors. Mol. Cell. Proteom. 2017, 16, S215–S229. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Pulit-Penaloza, J.A.; Scherbik, S.V.; Brinton, M.A. Type 1 IFN-independent activation of a subset of interferon stimulated genes in west nile virus Eg101-infected mouse cells. Virology 2012, 425, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, S.C.; Cho, H.; Chung, B.C.; Gale, M., Jr.; Chanda, S.K.; Diamond, M.S. A short hairpin RNA screen of interferon-stimulated genes identifies a novel negative regulator of the cellular antiviral response. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.B.; Miyauchi-Ishida, S.; Arimoto, K.; Liu, D.; Yan, M.; Liu, C.W.; Gyorffy, B.; Zhang, D.E. Type I IFN induces protein isgylation to enhance cytokine expression and augments colonic inflammation. Proc. Natl. Acad. Sci. USA 2015, 112, 14313–14318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Fu, F.; Xue, M.; Chen, W.; Liu, J.; Shi, H.; Chen, J.; Bu, Z.; Feng, L.; Liu, P. IFN-lambda preferably inhibits PEDV infection of porcine intestinal epithelial cells compared with IFN-alpha. Antivir. Res. 2017, 140, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.J.; Hahn, C.S.; Kim, K.I.; Yan, M.; Rosario, D.; Li, L.; de la Torre, J.C.; Zhang, D.E. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 2004, 10, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Ketscher, L.; Hannss, R.; Morales, D.J.; Basters, A.; Guerra, S.; Goldmann, T.; Hausmann, A.; Prinz, M.; Naumann, R.; Pekosz, A.; et al. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Hwang, S.Y.; Imaizumi, T.; Yoo, J.Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.R.; Monte, K.; Thackray, L.B.; Lenschow, D.J. ISG15 functions as an interferon-mediated antiviral effector early in the murine norovirus life cycle. J. Virol. 2014, 88, 9277–9286. [Google Scholar] [CrossRef] [PubMed]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, F.; Pal, S.; Sen, G.C. Induction and mode of action of the viral stress-inducible murine proteins, p56 and p54. Virology 2005, 340, 116–124. [Google Scholar] [CrossRef] [PubMed]
- McFadden, N.; Bailey, D.; Carrara, G.; Benson, A.; Chaudhry, Y.; Shortland, A.; Heeney, J.; Yarovinsky, F.; Simmonds, P.; Macdonald, A.; et al. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog. 2011, 7, e1002413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartour, K.; Nguyen, X.N.; Appourchaux, R.; Assil, S.; Barateau, V.; Bloyet, L.M.; Burlaud Gaillard, J.; Confort, M.P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of ifitms. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. Ifitm-family proteins: The cell’s first line of antiviral defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Everitt, A.R.; Clare, S.; McDonald, J.U.; Kane, L.; Harcourt, K.; Ahras, M.; Lall, A.; Hale, C.; Rodgers, A.; Young, D.B.; et al. Defining the range of pathogens susceptible to IFITM3 restriction using a knockout mouse model. PLoS ONE 2013, 8, e80723. [Google Scholar] [CrossRef] [PubMed]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 inhibits influenza a virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef] [PubMed]
- Anafu, A.A.; Bowen, C.H.; Chin, C.R.; Brass, A.L.; Holm, G.H. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J. Biol. Chem. 2013, 288, 17261–17271. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Griffin, L.M.; Little, A.S.; Huang, I.C.; Farzan, M.; Pyeon, D. The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS ONE 2014, 9, e96579. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Beard, M.R. The role of viperin in the innate antiviral response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hinson, E.R.; Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, X.; Wu, H.; Liu, C.; Li, Z.; Hu, X.; Su, S.; Wang, L.F.; Qu, L. Blocking the PI3K/AKT pathway enhances mammalian reovirus replication by repressing IFN-stimulated genes. Front. Microbiol. 2015, 6, 886. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Yaneva, R.; Hinson, E.R.; Cresswell, P. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 2011, 332, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Sharma, A.; Greenberg, H.B. Rotavirus degrades multiple type interferon receptors to inhibit ifn signaling and protects against mortality from endotoxin in suckling mice. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.; Ogden, K.M.; Patton, J.T. Silencing the alarms: Innate immune antagonism by rotavirus NSP1 and VP3. Virology 2015, 479–480, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Barro, M.; Patton, J.T. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 2007, 81, 4473–4481. [Google Scholar] [CrossRef] [PubMed]
- Barro, M.; Patton, J.T. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of ifn regulatory factor 3. Proc. Natl. Acad. Sci. USA 2005, 102, 4114–4119. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, P.; Bhowmick, R.; Nandi, S.; Kant Nayak, M.; Chawla-Sarkar, M. Rotavirus NSP1 inhibits interferon induced non-canonical NFκB activation by interacting with TNF receptor associated factor 2. Virology 2013, 444, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Ren, L.; Zhou, Z.; Lei, X.; Chen, L.; Xue, Q.; Liu, X.; Wang, J.; Hung, T. Rotavirus nonstructural protein 1 antagonizes innate immune response by interacting with retinoic acid inducible gene I. Virol. J. 2011, 8, 526. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Chanda, S.; Bagchi, P.; Nayak, M.K.; Bhowmick, R.; Chawla-Sarkar, M. MAVS protein is attenuated by rotavirus nonstructural protein 1. PLoS ONE 2014, 9, e92126. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.; Dennis, A.F.; Patton, J.T. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-κb activation by using molecular mimicry to target beta-TrCP. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Onoguchi, K.; Yoneyama, M.; Takemura, A.; Akira, S.; Taniguchi, T.; Namiki, H.; Fujita, T. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2007, 282, 7576–7581. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.; Dang, V.T.; Jans, D.A.; Coulson, B.S. Rotavirus inhibits IFN-induced stat nuclear translocation by a mechanism that acts after STAT binding to importin-alpha. J. Gen. Virol. 2014, 95, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Rott, L.; Phan, N.; Mukherjee, G.; Greenberg, H.B. Rotavirus NSP1 protein inhibits interferon-mediated STAT1 activation. J. Virol. 2014, 88, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H.; Weiss, S.R. Viral phosphodiesterases that antagonize double-stranded RNA signaling to RNase l by degrading 2-5A. J. Interferon Cytokine Res. 2014, 34, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Jha, B.K.; Ogden, K.M.; Dong, B.; Zhao, L.; Elliott, R.; Patton, J.T.; Silverman, R.H.; Weiss, S.R. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 13114–13119. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tacuba, L.; Rojas, M.; Arias, C.F.; Lopez, S. Rotavirus controls activation of the 2′-5′-oligoadenylate synthetase/RNase L pathway using at least two distinct mechanisms. J. Virol. 2015, 89, 12145–12153. [Google Scholar] [CrossRef] [PubMed]
- Kahan, S.M.; Liu, G.; Reinhard, M.K.; Hsu, C.C.; Livingston, R.S.; Karst, S.M. Comparative murine norovirus studies reveal a lack of correlation between intestinal virus titers and enteric pathology. Virology 2011, 421, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Kischnick, C.; Rippert, A.; Albrecht, D.; Boulant, S. Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci. Rep. 2017, 7, 10873. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.; Svedin, E.; Domsgen, E.; Kapell, S.; Laitinen, O.; Moll, M.; Flodstrom-Tullberg, M. Coxsackievirus counters the host innate immune response by blocking type III interferon expression. J. Gen. Virol. 2016, 97, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Rahbek, S.H.; Gad, H.H.; Bak, R.O.; Jakobsen, M.R.; Jiang, Z.; Hansen, A.L.; Jensen, S.K.; Sun, C.; Thomsen, M.K.; et al. Influenza a virus targets a CGAS-independent sting pathway that controls enveloped rna viruses. Nat. Commun. 2016, 7, 10680. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wu, Y.; Zhang, W.; Qi, J.; Gao, G.F. Enabling the ‘host jump’: Structural determinants of receptor-binding specificity in influenza a viruses. Nat. Rev. Microbiol. 2014, 12, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Jensen, S.B.; Jakobsen, M.R.; Cheshenko, N.; Horan, K.A.; Moeller, H.B.; Gonzalez-Dosal, R.; Rasmussen, S.B.; Christensen, M.H.; Yarovinsky, T.O.; et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor sting. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Interferon induction and function at the mucosal surface. Immunol. Rev. 2013, 255, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune functions at barrier surfaces and beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Wack, A.; Terczynska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Marchiando, A.M.; Graham, W.V.; Turner, J.R. Epithelial barriers in homeostasis and disease. Annu. Rev. Pathol. 2010, 5, 119–144. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, V.; Dutta, O.; McElrath, C.; Du, P.; Chang, Y.J.; Cicciarelli, B.; Pitler, A.; Whitehead, I.; Obar, J.J.; Durbin, J.E.; et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Katakura, K.; Lee, J.; Rachmilewitz, D.; Li, G.; Eckmann, L.; Raz, E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Investig. 2005, 115, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Lienenklaus, S.; Cornitescu, M.; Zietara, N.; Lyszkiewicz, M.; Gekara, N.; Jablonska, J.; Edenhofer, F.; Rajewsky, K.; Bruder, D.; Hafner, M.; et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J. Immunol. 2009, 183, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Kotredes, K.P.; Thomas, B.; Gamero, A.M. The protective role of type I interferons in the gastrointestinal tract. Front. Immunol. 2017, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Tschurtschenthaler, M.; Wang, J.; Fricke, C.; Fritz, T.M.; Niederreiter, L.; Adolph, T.E.; Sarcevic, E.; Kunzel, S.; Offner, F.A.; Kalinke, U.; et al. Type I interferon signalling in the intestinal epithelium affects paneth cells, microbial ecology and epithelial regeneration. Gut 2014, 63, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Hainzl, E.; Rosebrock, F.; Heider, S.; Schwab, C.; Berry, D.; Stoiber, D.; Wagner, M.; Schleper, C.; Loy, A.; et al. Type I interferons have opposing effects during the emergence and recovery phases of colitis. Eur. J. Immunol. 2014, 44, 2749–2760. [Google Scholar] [CrossRef] [PubMed]
- Katlinskaya, Y.V.; Katlinski, K.V.; Lasri, A.; Li, N.; Beiting, D.P.; Durham, A.C.; Yang, T.; Pikarsky, E.; Lengner, C.J.; Johnson, F.B.; et al. Type I interferons control proliferation and function of the intestinal epithelium. Mol. Cell. Biol. 2016, 36, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Katlinskaya, Y.V.; Carbone, C.J.; Zhao, B.; Katlinski, K.V.; Zheng, H.; Guha, M.; Li, N.; Chen, Q.; Yang, T.; et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Robb, R.J.; Kreijveld, E.; Kuns, R.D.; Wilson, Y.A.; Olver, S.D.; Don, A.L.; Raffelt, N.C.; De Weerd, N.A.; Lineburg, K.E.; Varelias, A.; et al. Type I-IFNs control GVHD and GVL responses after transplantation. Blood 2011, 118, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.C.; Bscheider, M.; Eisenkolb, G.; Lin, C.C.; Wintges, A.; Otten, V.; Lindemans, C.A.; Heidegger, S.; Rudelius, M.; Monette, S.; et al. RIG-I/MAVS and sting signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Nguyen, K.P.; Fine, S.D.; Mo, J.H.; Shen, C.; Shenouda, S.; Corr, M.; Jung, S.; Lee, J.; Eckmann, L.; et al. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 17022–17027. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Chiriac, M.T.; Buchen, B.; Wandersee, A.; Hundorfean, G.; Gunther, C.; Bourjau, Y.; Doyle, S.E.; Frey, B.; Ekici, A.B.; Buttner, C.; et al. Activation of epithelial STAT1 by interleukin 28 controls mucosal healing in mice with colitis and is increased in mucosa of patients with inflammatory bowel disease. Gastroenterology 2017, 153, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Rosebrock, F.; Hainzl, E.; Heider, S.; Majoros, A.; Wienerroither, S.; Strobl, B.; Stockinger, S.; Kenner, L.; Muller, M.; et al. Noncanonical effects of IRF9 in intestinal inflammation: More than type I and type III interferons. Mol. Cell. Biol. 2015, 35, 2332–2343. [Google Scholar] [CrossRef] [PubMed]
- Broggi, A.; Tan, Y.; Granucci, F.; Zanoni, I. IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat. Immunol. 2017, 18, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [Green Version]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded human microbiome project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Tropini, C.; Earle, K.A.; Huang, K.C.; Sonnenburg, J.L. The gut microbiome: Connecting spatial organization to function. Cell Host Microbe 2017, 21, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, R.; Chassaing, B.; Zhang, B.; Gewirtz, A.T. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J. Infect. Dis. 2014, 210, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza a virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef] [PubMed]
- Steed, A.L.; Christophi, G.P.; Kaiko, G.E.; Sun, L.; Goodwin, V.M.; Jain, U.; Esaulova, E.; Artyomov, M.N.; Morales, D.J.; Holtzman, M.J.; et al. The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science 2017, 357, 498–502. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, A.J.; McSorley, H.J.; Davidson, D.J.; Fitch, P.M.; Errington, C.; Mackenzie, K.J.; Gollwitzer, E.S.; Johnston, C.J.C.; MacDonald, A.S.; Edwards, M.R.; et al. Enteric helminth-induced type I interferon signaling protects against pulmonary virus infection through interaction with the microbiota. J. Allergy Clin. Immunol. 2017, 140, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Miller, H.L.; Sesti-Costa, R.; Cella, M.; Gilfillan, S.; Colonna, M. Microbiota induces tonic CCL2 systemic levels that control pdc trafficking in steady state. Mucosal Immunol. 2017, 10, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Kim, M.S.; Kim, E.; Cheon, J.H.; Lee, Y.S.; Kim, Y.; Lee, S.H.; Seo, S.U.; Shin, S.H.; Choi, S.S.; et al. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-beta production. Immunity 2016, 44, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, S.; Yang, L.; Cui, S.; Pan, W.; Jackson, R.; Zheng, Y.; Rongvaux, A.; Sun, Q.; Yang, G.; et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 2015, 350, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Planet, P.J.; Parker, D.; Cohen, T.S.; Smith, H.; Leon, J.D.; Ryan, C.; Hammer, T.J.; Fierer, N.; Chen, E.I.; Prince, A.S. Lambda interferon restructures the nasal microbiome and increases susceptibility to staphylococcus aureus superinfection. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.M.; Elftman, M.D.; Pinto, A.K.; Baldridge, M.; Hooper, P.; Kuczynski, J.; Petrosino, J.F.; Young, V.B.; Wobus, C.E. Murine norovirus infection does not cause major disruptions in the murine intestinal microbiota. Microbiome 2013, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Hickman, D.; Jones, M.K.; Zhu, S.; Kirkpatrick, E.; Ostrov, D.A.; Wang, X.; Ukhanova, M.; Sun, Y.; Mai, V.; Salemi, M.; et al. The effect of malnutrition on norovirus infection. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.M.; Walk, S.T.; Taube, S.; Taniuchi, M.; Houpt, E.R.; Wobus, C.E.; Young, V.B. Disruption of the human gut microbiota following norovirus infection. PLoS ONE 2012, 7, e48224. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Tsai, C.N.; Lee, Y.S.; Lin, C.Y.; Huang, K.Y.; Chao, H.C.; Lai, M.W.; Chiu, C.H. Intestinal microbiome in children with severe and complicated acute viral gastroenteritis. Sci. Rep. 2017, 7, 46130. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 2017, 46, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Hartmann, R.; Wack, A. IFNlambda is a potent anti-influenza therapeutic without the inflammatory side effects of IFNalpha treatment. EMBO Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika virus and birth defects—Reviewing the evidence for causality. N. Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika virus associated with microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A mouse model of zika virus pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.W.; Woods, T.A.; Rosenke, R.; Scott, D.P.; Best, S.M.; Peterson, K.E. Sexual and vertical transmission of zika virus in anti-interferon receptor-treated RAG1-deficient mice. Sci. Rep. 2017, 7, 7176. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liang, Y.; Yi, P.; Xu, L.; Hawkins, H.K.; Rossi, S.L.; Soong, L.; Cai, J.; Menon, R.; Sun, J. Outcomes of congenital zika disease depend on timing of infection and maternal-fetal interferon action. Cell Rep. 2017, 21, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III interferons produced by human placental trophoblasts confer protection against zika virus infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Corry, J.; Arora, N.; Good, C.A.; Sadovsky, Y.; Coyne, C.B. Organotypic models of type III interferon-mediated protection from zika virus infections at the maternal-fetal interface. Proc. Natl. Acad. Sci. USA 2017, 114, 9433–9438. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Daniels, B.P.; Pinto, A.K.; Huang, A.C.; Vick, S.C.; Doyle, S.E.; Gale, M., Jr.; Klein, R.S.; Diamond, M.S. Interferon-lambda restricts west nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl. Med. 2015, 7, 284ra259. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal west nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Brassil, M.M.; Blahnik, G.; McMillan, A.; Ramos, H.J.; Proll, S.C.; Belisle, S.E.; Katze, M.G.; Gale, M., Jr. A systems biology approach reveals that tissue tropism to west nile virus is regulated by antiviral genes and innate immune cellular processes. PLoS Pathog. 2013, 9, e1003168. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.K.; Ramos, H.J.; Wu, X.; Aggarwal, S.; Shrestha, B.; Gorman, M.; Kim, K.Y.; Suthar, M.S.; Atkinson, J.P.; Gale, M., Jr.; et al. Deficient IFN signaling by myeloid cells leads to mavs-dependent virus-induced sepsis. PLoS Pathog. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Soto Albrecht, Y.E.; Hrebikova, G.; Sadimin, E.; Davidson, C.; Kotenko, S.V.; Ploss, A. Type III interferon-mediated signaling is critical for controlling live attenuated yellow fever virus infection in vivo. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.J.; Shiffman, M.L.; Zaman, A.; Yoffe, B.; de la Torre, A.; Flamm, S.; Gordon, S.C.; Marotta, P.; Vierling, J.M.; Lopez-Talavera, J.C.; et al. Phase 1B study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology 2010, 52, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.C.; Armah, G.; Fuentes, S.; Korpela, K.E.; Parashar, U.; Victor, J.C.; Tate, J.; de Weerth, C.; Giaquinto, C.; Wiersinga, W.J.; et al. Significant correlation between the infant gut microbiome and rotavirus vaccine response in rural ghana. J. Infect. Dis. 2017, 215, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.; Ali, A.; Fuentes, S.; Korpela, K.; Kazi, M.; Tate, J.; Parashar, U.; Wiersinga, W.J.; Giaquinto, C.; de Weerth, C.; et al. Rotavirus vaccine response correlates with the infant gut microbiota composition in pakistan. Gut Microbes 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Type I IFN | Type III IFN | References |
|---|---|---|---|
| Norovirus (NoV) |
|
| [45,52,54,56,58,61,62,63] |
| Reovirus |
|
| [45,48,64,65] |
| Rotavirus (RV) |
|
| [27,47,66,67,68,69] |
| Adenovirus (AdV) |
|
| [70,71,72] |
| Murine cytomegalovirus (MCMV) |
|
| [73,74] |
| Challenge | Type I IFN | Type III IFN | References |
|---|---|---|---|
| Inflammation and colitis |
|
| [140,144,147,148,149,151,152,153] |
| Injury repair |
|
| [86,145,146,151] |
| Homeostasis |
|
| [143,151] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ingle, H.; Peterson, S.T.; Baldridge, M.T. Distinct Effects of Type I and III Interferons on Enteric Viruses. Viruses 2018, 10, 46. https://doi.org/10.3390/v10010046
Ingle H, Peterson ST, Baldridge MT. Distinct Effects of Type I and III Interferons on Enteric Viruses. Viruses. 2018; 10(1):46. https://doi.org/10.3390/v10010046
Chicago/Turabian StyleIngle, Harshad, Stefan T. Peterson, and Megan T. Baldridge. 2018. "Distinct Effects of Type I and III Interferons on Enteric Viruses" Viruses 10, no. 1: 46. https://doi.org/10.3390/v10010046
APA StyleIngle, H., Peterson, S. T., & Baldridge, M. T. (2018). Distinct Effects of Type I and III Interferons on Enteric Viruses. Viruses, 10(1), 46. https://doi.org/10.3390/v10010046