Isolation of A Novel Bacillus thuringiensis Phage Representing A New Phage Lineage and Characterization of Its Endolysin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Phage Isolation, Purification, and Observation
2.3. Bacterial and Phage Genomic DNA Purification, Genome Sequencing, and Bioinformatic Analysis
2.4. Expression and Purification of the Endolysin PlyBMB
2.5. Lytic Activity Assay of PlyBMB
2.6. GenBank Accession Number
3. Results
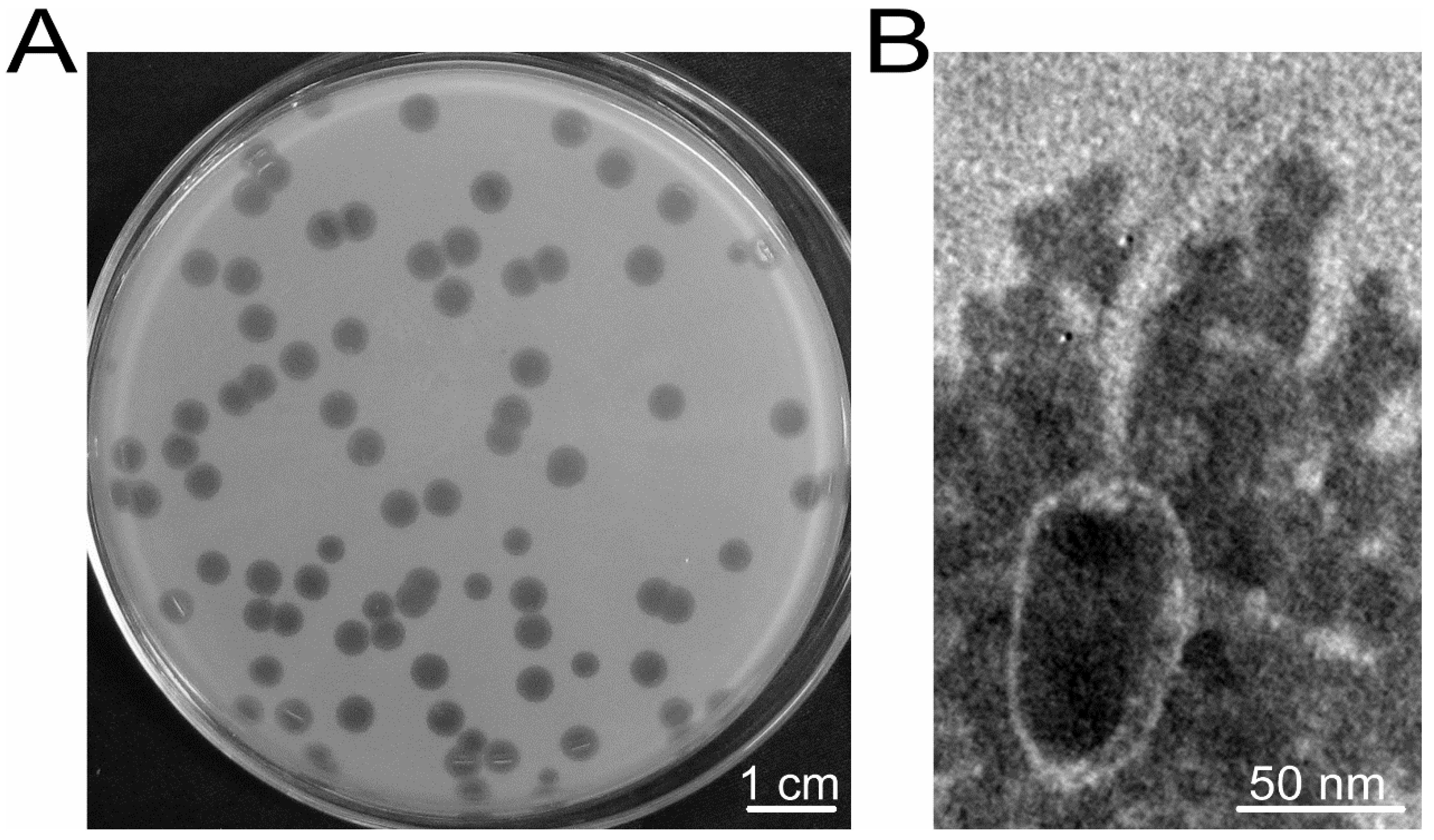
3.1. Phage vB_BthS_BMBphi Exhibits High Lytic Activity and High Specificity
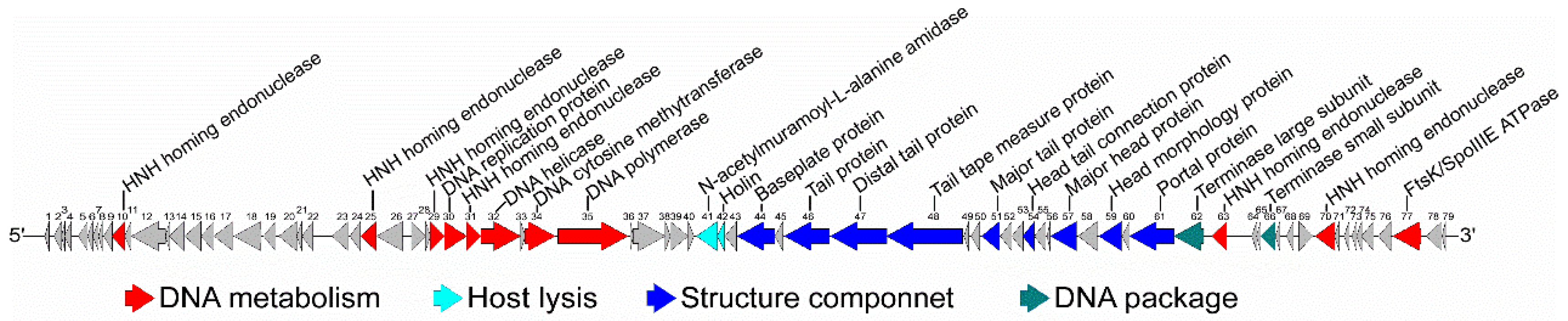
3.2. General Genomic Characteristics of the Phage vB_BthS_BMBphi
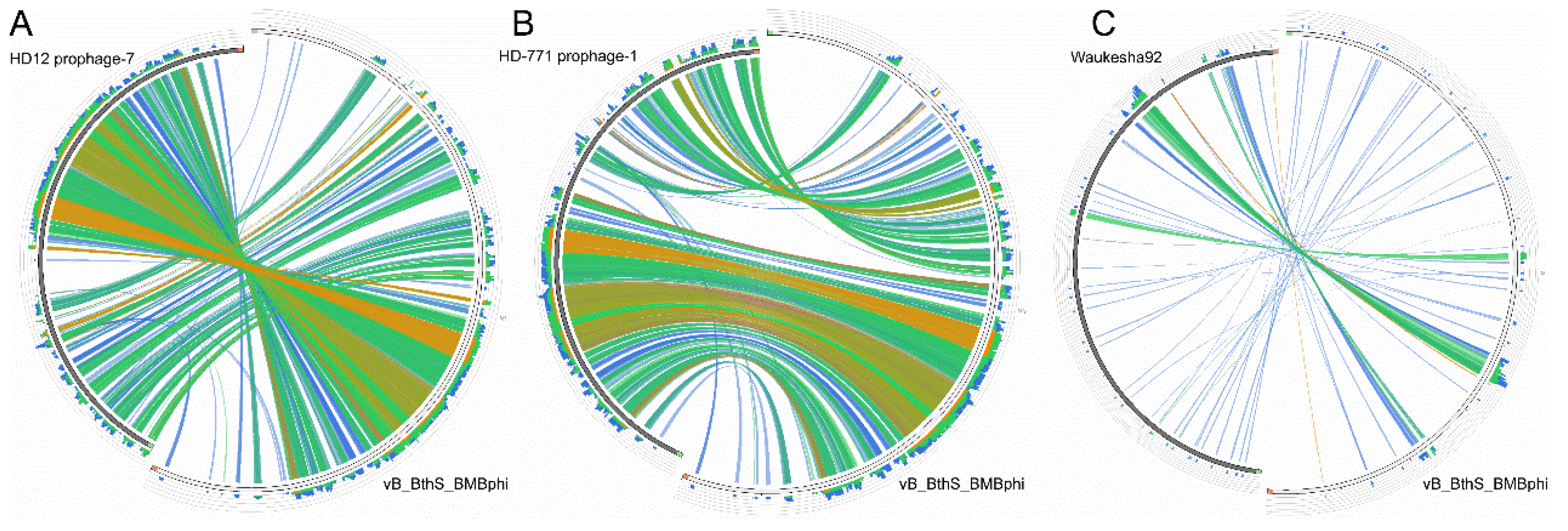
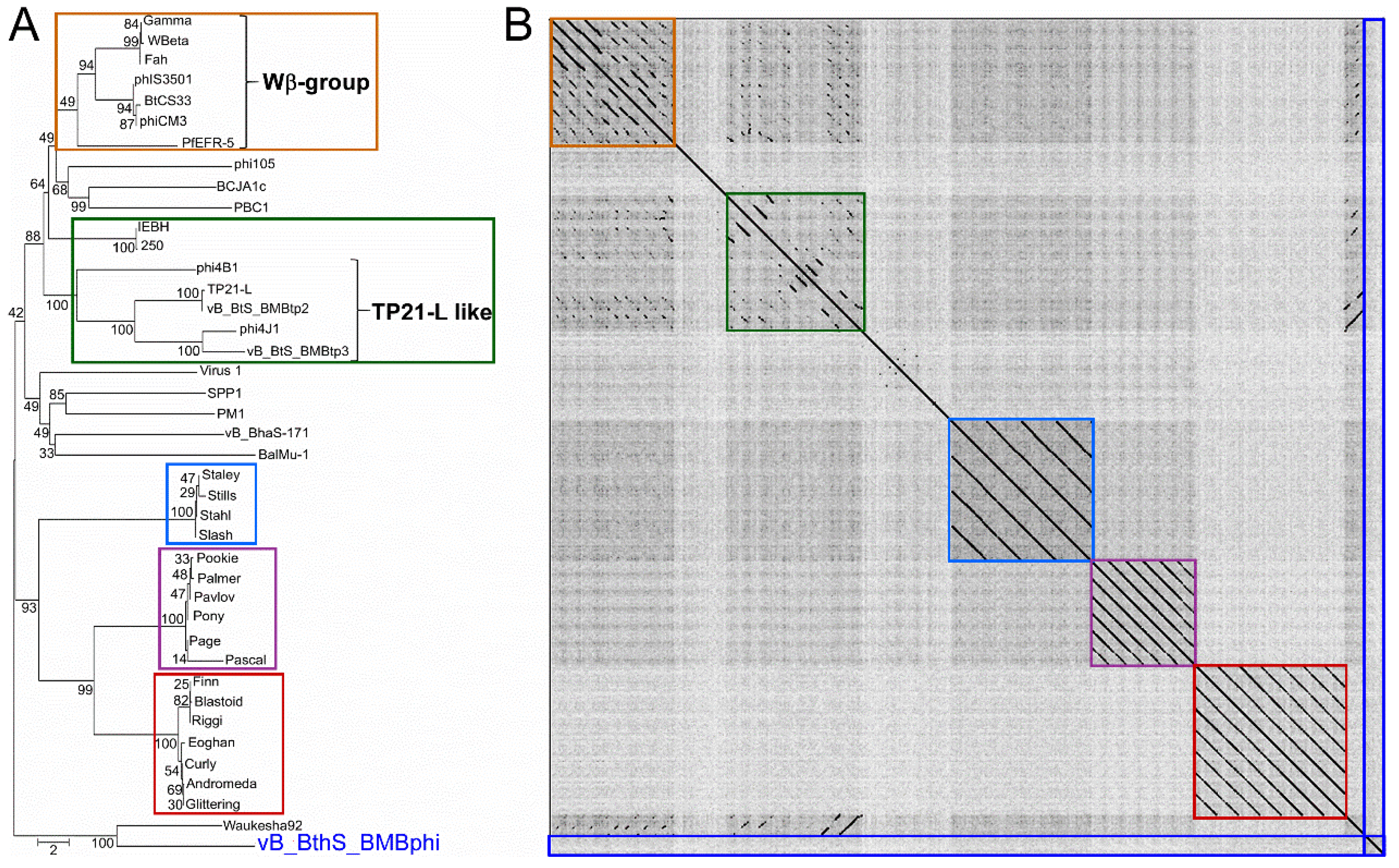
3.3. The Phage vB_BthS_BMBphi Contains a Novel Genome
3.4. The Phage vB_BthS_BMBphi Genome Contains a Repeated Terminal Structure
3.5. Lytic Activity of the Endolysin PlyBMB
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rohwer, F. Global phage diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef]
- Yen, M.; Cairns, L.S.; Camilli, A. A cocktail of three virulent bacteriophages prevents Vibrio cholerae infection in animal models. Nat. Commun. 2017, 8, 14187. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, F.L.; Costa, A.R.; Kluskens, L.D.; Azeredo, J. Revisiting phage therapy: New applications for old resources. Trends Microbiol. 2015, 23, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, C.; Venail, P.; Holguin, A.V.; Vives, M.J. Co-Evolutionary Dynamics of the Bacteria Vibrio sp. CV1 and Phages V1G, V1P1, and V1P2: Implications for Phage Therapy. Microb. Ecol. 2013, 66, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a Broad-Range Bacteriophage Cocktail That Reduces Pseudomonas aeruginosa Biofilms and Treats Acute Infections in Two Animal Models. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Pirnay, J.P.; de Vos, D. Guidelines to Compose an Ideal Bacteriophage Cocktail. Methods Mol. Biol. 2018, 1693, 99–110. [Google Scholar] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Schnepf, E.; Crickmore, N.; van Rie, J.; Lereclus, D.; Baum, J.; Feitelson, J.; Zeigler, D.R.; Dean, D.H. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 1998, 62, 775–806. [Google Scholar] [PubMed]
- Vilas-Boas, G.T.; Peruca, A.P.; Arantes, O.M. Biology and taxonomy of Bacillus cereus, Bacillus anthracis, and Bacillus thuringiensis. Can. J. Microbiol. 2007, 53, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Bottone, E.J. Bacillus cereus, a volatile human pathogen. Clin. Microbiol. Rev. 2010, 23, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Koehler, T.M. Bacillus anthracis genetics and virulence gene regulation. Curr. Top. Microbiol. Immunol. 2002, 271, 143–164. [Google Scholar] [PubMed]
- Schuch, R.; Fischetti, V.A. Detailed genomic analysis of the Wbeta and gamma phages infecting Bacillus anthracis: Implications for evolution of environmental fitness and antibiotic resistance. J. Bacteriol. 2006, 188, 3037–3051. [Google Scholar] [CrossRef] [PubMed]
- Nakonieczna, A.; Cooper, C.J.; Gryko, R. Bacteriophages and bacteriophage-derived endolysins as potential therapeutics to combat Gram-positive spore forming bacteria. J. Appl. Microbiol. 2015, 119, 620–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Shao, X.; Zheng, H.; Li, M.; Wang, J.; Zhang, Q.; Li, L.; Liu, Z.; Sun, M.; Wang, S.; et al. Complete genome sequence of Bacillus thuringiensis mutant strain BMB171. J. Bacteriol. 2010, 192, 4074–4075. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Yuan, Y.H.; Gao, M.Y. Construction of an environmental safe Bacillus thuringiensis engineered strain against Coleoptera. Appl. Microbiol. Biotechnol. 2016, 100, 4027–4034. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.H.; Peng, Q.; Wu, D.D.; Kou, Z.; Wu, Y.; Liu, P.M.; Gao, M.Y. Effects of Actin-Like Proteins Encoded by Two Bacillus pumilus Phages on Unstable Lysogeny, Revealed by Genomic Analysis. Appl. Environ. Microb. 2015, 81, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Hardies, S.C.; Rolando, M.; Hayes, S.J.; Lieman, K.; Carroll, C.A.; Weintraub, S.T.; Serwer, P. Complete genomic sequence and mass spectrometric analysis of highly diverse, atypical Bacillus thuringiensis phage 0305 phi 8-36. Virology 2007, 368, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.H.; Gao, M.Y. Genomic analysis of a ginger pathogen Bacillus pumilus providing the understanding to the pathogenesis and the novel control strategy. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- BLASTP. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 18 October 2018).
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life v2: Online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011, 39, W475–W478. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Thiagarajan, V.; Walmagh, M.; Sillankorva, S.; Lavigne, R.; Neves-Petersen, M.T.; Kluskens, L.D.; Azeredo, J. A thermostable Salmonella phage endolysin, Lys68, with broad bactericidal properties against gram-negative pathogens in presence of weak acids. PLoS ONE 2014, 9, e108376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walmagh, M.; Boczkowska, B.; Grymonprez, B.; Briers, Y.; Drulis-Kawa, Z.; Lavigne, R. Characterization of five novel endolysins from Gram-negative infecting bacteriophages. Appl. Microbiol. Biotechnol. 2013, 97, 4369–4375. [Google Scholar] [CrossRef] [PubMed]
- Dziewit, L.; Oscik, K.; Bartosik, D.; Radlinska, M. Molecular Characterization of a Novel Temperate Sinorhizobium Bacteriophage, Phi LM21, Encoding DNA Methyltransferase with CcrM-Like Specificity. J. Virol. 2014, 88, 13111–13124. [Google Scholar] [CrossRef] [PubMed]
- Dziewit, L.; Radlinska, M. Two novel temperate bacteriophages co-existing in Aeromonas sp. ARM81-characterization of their genomes, proteomes and DNA methyltransferases. J. Gen. Virol. 2016, 97, 2008–2022. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE-a database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef] [PubMed]
- Crutz-Le Coq, A.M.; Cesselin, B.; Commissaire, J.; Anba, J. Sequence analysis of the lactococcal bacteriophage bIL170: Insights into structural proteins and HNH endonucleases in dairy phages. Microbiology 2002, 148, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, B.S.; Stoddard, B.L. Homing endonucleases: Structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001, 29, 3757–3774. [Google Scholar] [CrossRef] [PubMed]
- Golz, S.; Kemper, B. Association of holliday-structure resolving endonuclease VII with gp20 from the packaging machine of phage T4. J. Mol. Biol. 1999, 285, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Walter, M.H.; Yang, L.B.; Chen, S.C.; Winston, V.; Thomas, M.A. Predicting genome terminus sequences of Bacillus cereus-group bacteriophage using next generation sequencing data. BMC Genom. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Etobayeva, I.; Linden, S.B.; Alem, F.; Harb, L.; Rizkalla, L.; Mosier, P.D.; Johnson, A.A.; Temple, L.; Hakami, R.M.; Nelson, D.C. Discovery and Biochemical Characterization of PlyP56, PlyN74, and PlyTB40-Bacillus Specific Endolysins. Viruses 2018, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rubio, L.; Gutierrez, D.; Donovan, D.M.; Martinez, B.; Rodriguez, A.; Garcia, P. Phage lytic proteins: Biotechnological applications beyond clinical antimicrobials. Crit. Rev. Biotechnol. 2016, 36, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Loessner, M.J. Bacteriophage endolysins: Applications for food safety. Curr. Opin. Biotechnol. 2016, 37, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, O.; Rodriguez-Verdugo, A.; Gaut, R.L.; McDonald, P.; Bennett, A.F.; Long, A.D.; Gaut, B.S. The Molecular Diversity of Adaptive Convergence. Science 2012, 335, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gao, M.; Peng, Q.; Wu, D.; Liu, P.; Wu, Y. Genomic analysis of a phage and prophage from a Bacillus thuringiensis strain. J. Gen. Virol. 2014, 95, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Schmelcher, M.; Rodriguez-Rubio, L.; Klumpp, J.; Pritchard, D.G.; Dong, S.; Donovan, D.M. Endolysins as antimicrobials. Adv. Virus Res. 2012, 83, 299–365. [Google Scholar] [PubMed]
- Fernandes, S.; Proenca, D.; Cantante, C.; Silva, F.A.; Leandro, C.; Lourenco, S.; Milheirico, C.; de Lencastre, H.; Cavaco-Silva, P.; Pimentel, M.; et al. Novel chimerical endolysins with broad antimicrobial activity against methicillin-resistant Staphylococcus aureus. Microb. Drug Resist. 2012, 18, 333–343. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Peng, Q.; Yang, S.; Zhang, S.; Fu, Y.; Wu, Y.; Gao, M. Isolation of A Novel Bacillus thuringiensis Phage Representing A New Phage Lineage and Characterization of Its Endolysin. Viruses 2018, 10, 611. https://doi.org/10.3390/v10110611
Yuan Y, Peng Q, Yang S, Zhang S, Fu Y, Wu Y, Gao M. Isolation of A Novel Bacillus thuringiensis Phage Representing A New Phage Lineage and Characterization of Its Endolysin. Viruses. 2018; 10(11):611. https://doi.org/10.3390/v10110611
Chicago/Turabian StyleYuan, Yihui, Qin Peng, Shuo Yang, Shaowen Zhang, Yajuan Fu, Yan Wu, and Meiying Gao. 2018. "Isolation of A Novel Bacillus thuringiensis Phage Representing A New Phage Lineage and Characterization of Its Endolysin" Viruses 10, no. 11: 611. https://doi.org/10.3390/v10110611
APA StyleYuan, Y., Peng, Q., Yang, S., Zhang, S., Fu, Y., Wu, Y., & Gao, M. (2018). Isolation of A Novel Bacillus thuringiensis Phage Representing A New Phage Lineage and Characterization of Its Endolysin. Viruses, 10(11), 611. https://doi.org/10.3390/v10110611