Genomic Analysis of the Recent Viral Isolate vB_BthP-Goe4 Reveals Increased Diversity of φ29-Like Phages



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage Isolation and Genome Sequencing
2.2. Transmission Electron Microscopy
2.3. Genome Analysis and Comparison
2.4. Orthology and Evolutionary Analyses
3. Results
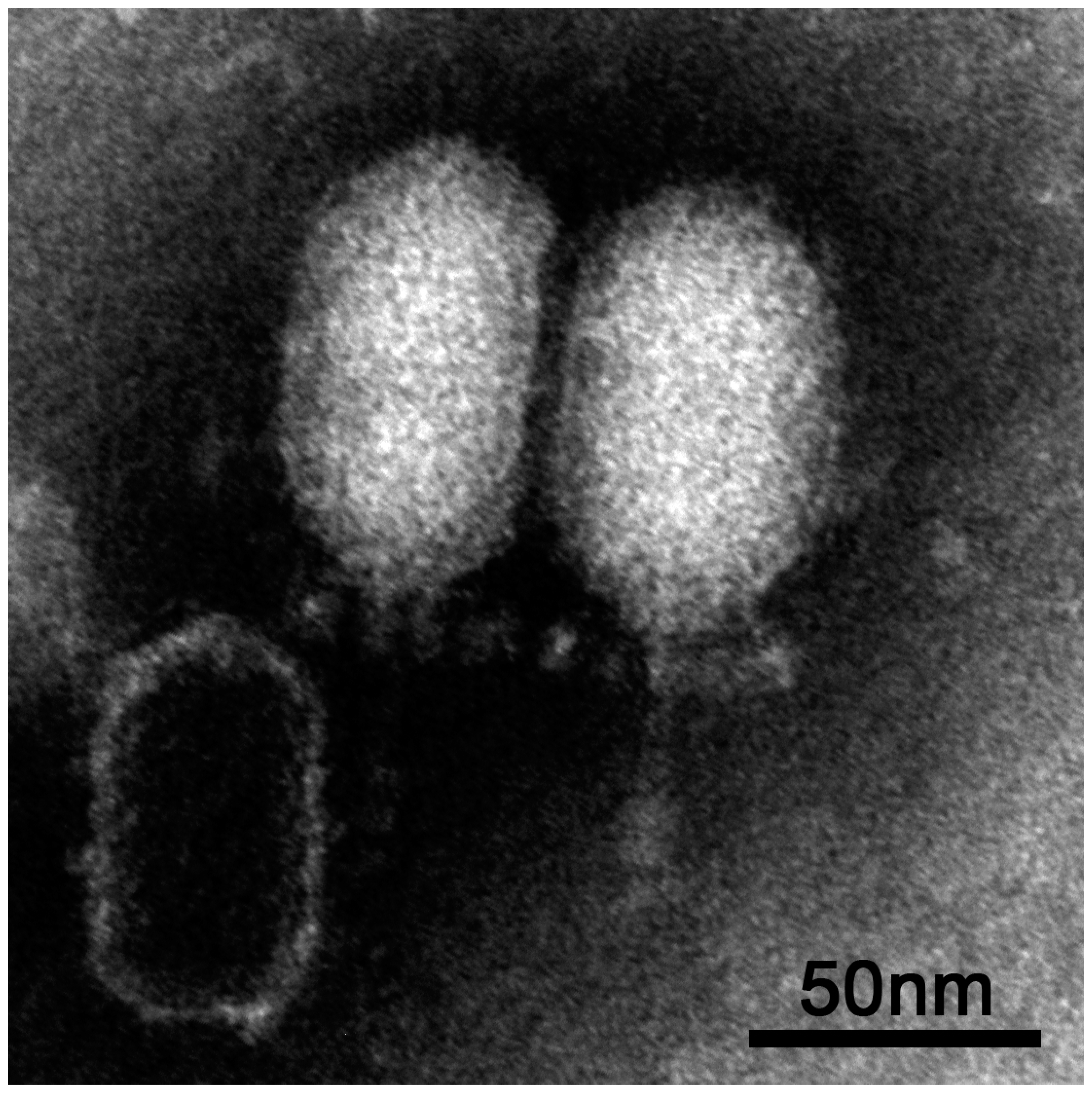
3.1. Isolation and Morphological Characterisation of vB_BthP-Goe4
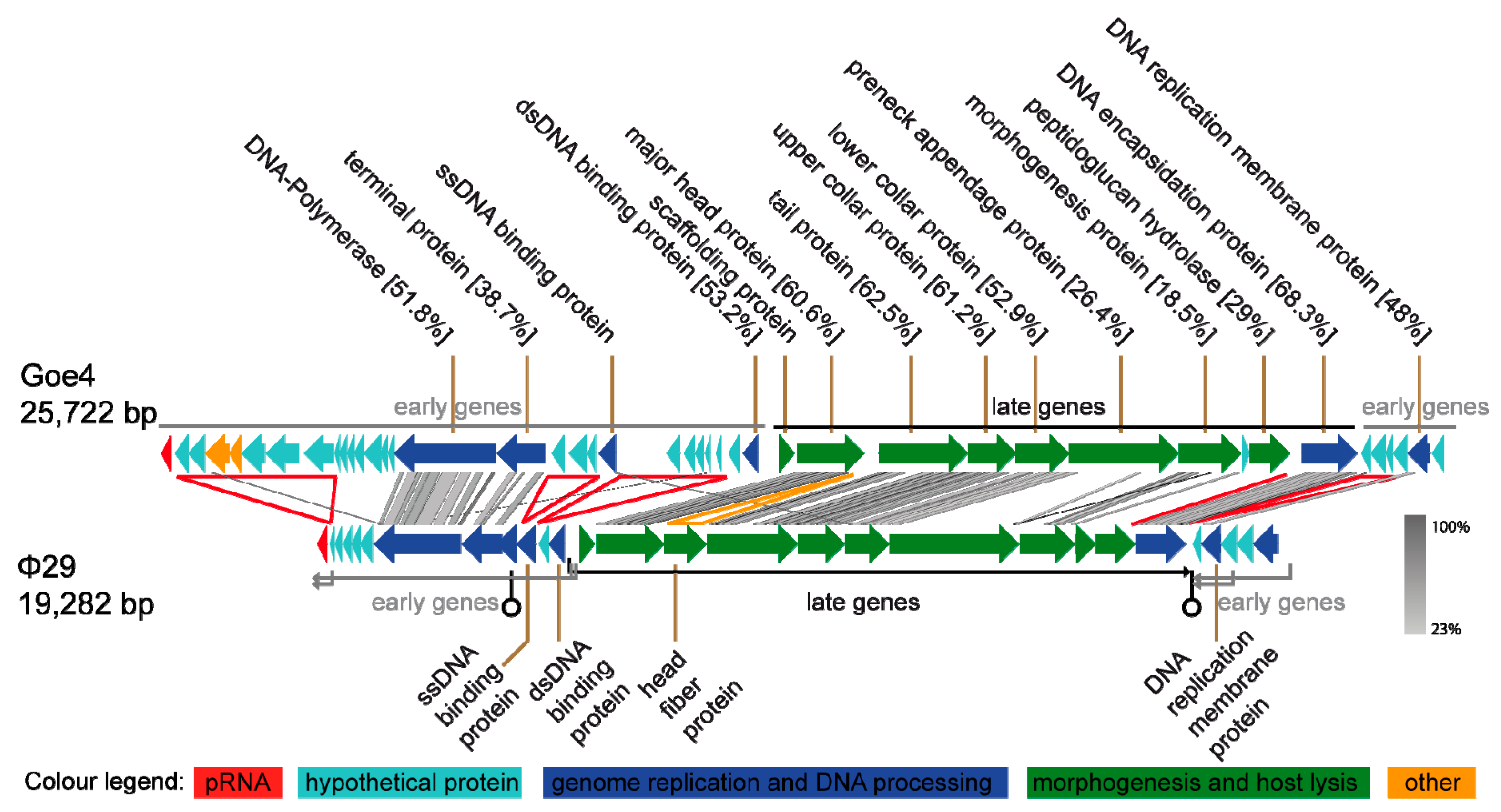
3.2. Genomic Characterization of vB_BthP-Goe4
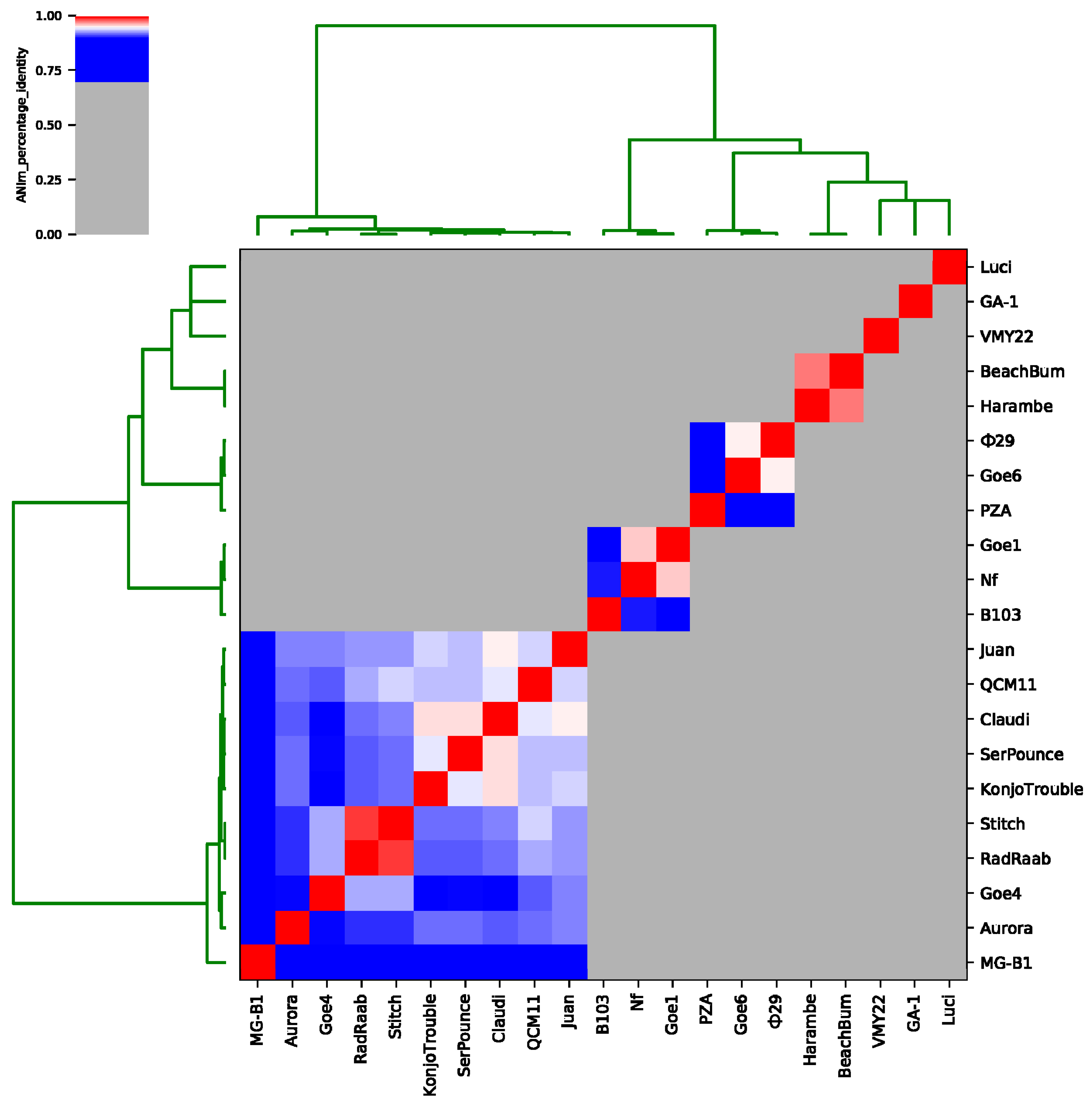
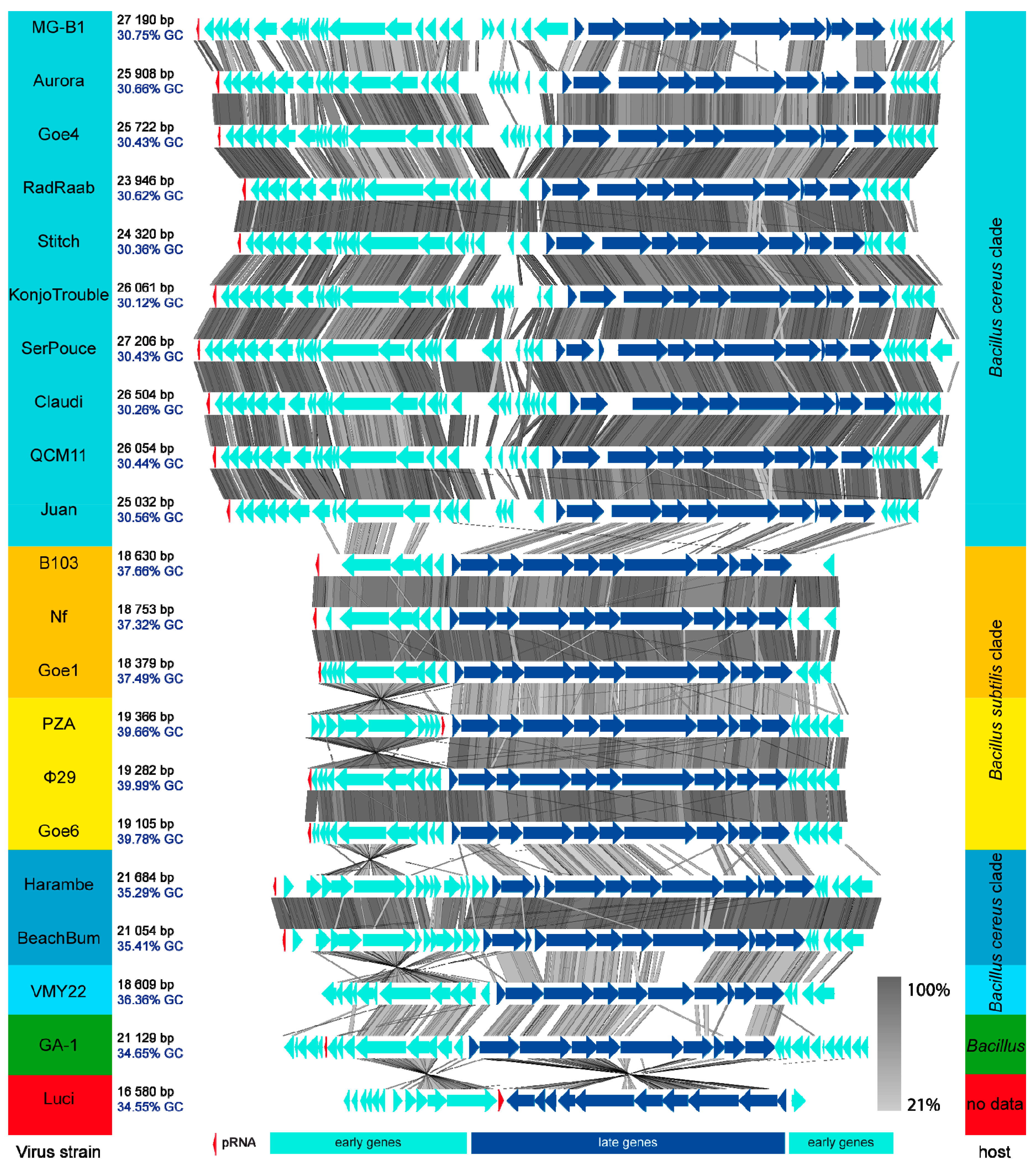
3.3. Goe4 and Its Closest Relatives
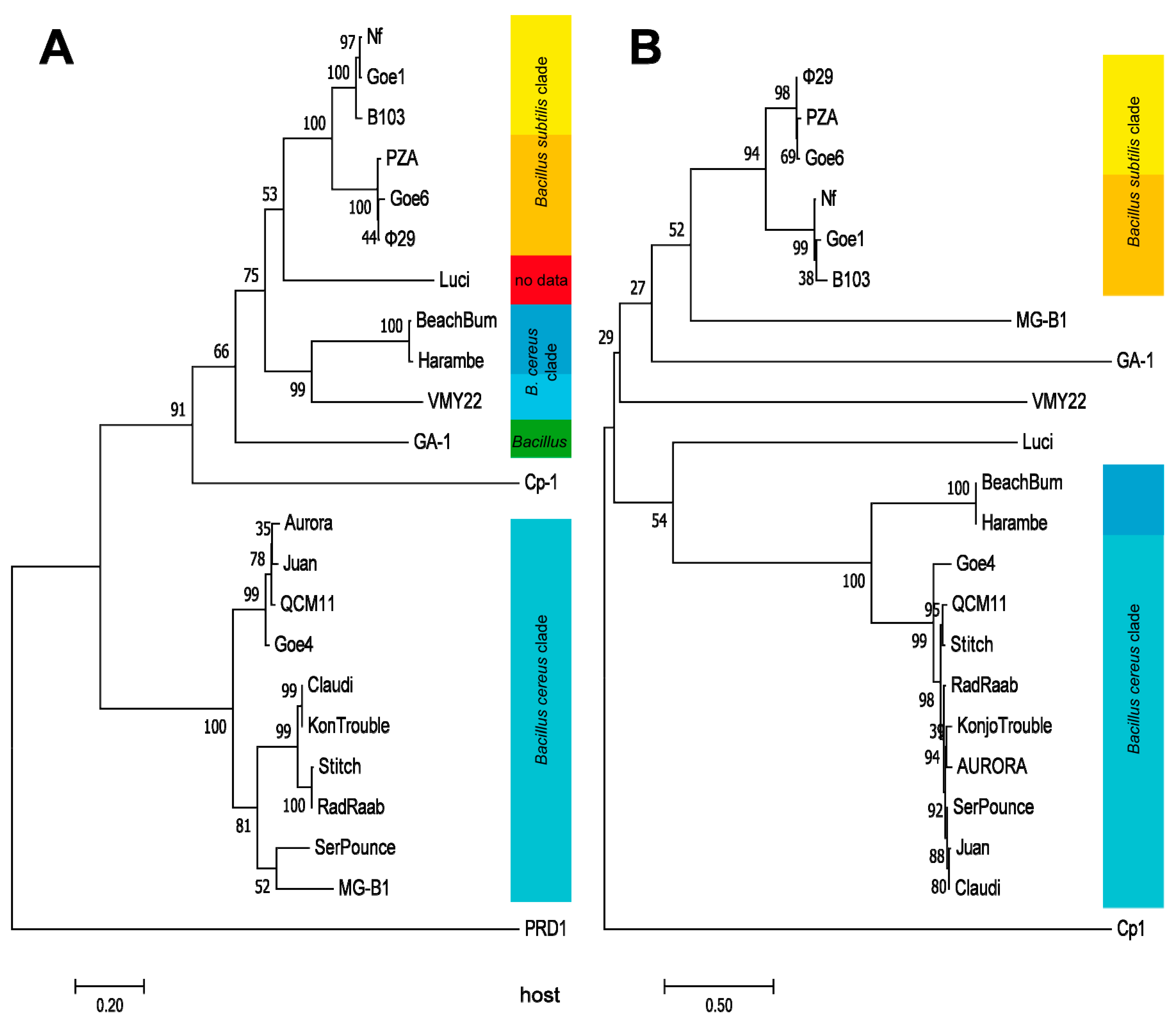
3.4. Orthology and Evolutionary Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Casjens, S.R. Comparative genomics and evolution of the tailed-bacteriophages. Curr. Opin. Microbiol. 2005, 8, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boodman, E. First Phage Therapy Center in the U.S. Signals Growing Acceptance. Available online: https://www.statnews.com/2018/06/21/first-phage-therapy-center-in-us/ (accessed on 19 August 2018).
- Logan, N.A.; De Vos, P. Systematic Bacteriology; Whitman, W.B., Ed.; Springer: New York, NY, USA, 2009; ISBN 978-0-387-95041-9. [Google Scholar]
- Hedges, S.B. The origin and evolution of model organisms. Nat. Rev. Genet. 2002, 3, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Calendar, R. The Bacteriophages; Oxford University Press: Oxford, UK, 2006; ISBN 9780195148503. [Google Scholar]
- Meijer, W.J.J.; Horcajadas, J.A.; Salas, M. phi29 Family of Phages. Microbiol. Mol. Biol. Rev. 2001, 65, 261–287. [Google Scholar] [CrossRef] [PubMed]
- Willms, I.M.; Hertel, R. Phage vB_BsuP-Goe1: The smallest identified lytic phage of Bacillus subtilis. FEMS Microbiol. Lett. 2016, 363, fnw208. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Lin, M.; Beroukhim, R.; Lee, J.C.; Zhao, X.; Richter, D.J.; Gabriel, S.; Herman, P.; Sasaki, H.; Altshuler, D.; et al. Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 2004, 32, e71. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.A.; Tao, Y.; Leiman, P.G.; Badasso, M.O.; He, Y.; Jardine, P.J.; Olson, N.H.; Morais, M.C.; Grimes, S.; Anderson, D.L.; et al. Structure of the bacteriophage phi29 DNA packaging motor. Nature 2000, 408, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Heras, G.; Bravo, A.; Salas, M. Phage phi29 protein p56 prevents viral DNA replication impairment caused by uracil excision activity of uracil-DNA glycosylase. Proc. Natl. Acad. Sci. USA 2008, 105, 19044–19049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Daniel, R.; Kawai, Y.; Carballido-López, R.; Castilla-Llorente, V.; Errington, J.; Meijer, W.J.J.; Salas, M. The actin-like MreB cytoskeleton organizes viral DNA replication in bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 13347–13352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/files/proposals/taxonomy_proposals_prokaryote1/m/bact04/7689 (accessed on 5 November 2018).
- Hendriksen, N.B.; Hansen, B.M. Detection of Bacillus thuringiensis kurstaki HD1 on cabbage for human consumption. FEMS Microbiol. Lett. 2006, 257, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Willms, I.M.; Hoppert, M.; Hertel, R. Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe3. Viruses 2017, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 November 2018).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Hertel, R.; Pintor Rodríguez, D.; Hollensteiner, J.; Dietrich, S.; Leimbach, A.; Hoppert, M.; Liesegang, H.; Volland, S. Genome-Based Identification of Active Prophage Regions by Next Generation Sequencing in Bacillus licheniformis DSM13. PLoS ONE 2015, 10, e0120759. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Doeven, M.K.; Kok, J.; Poolman, B.; Kuipers, O.P.; Kok, J.; Kuipers, O.; Chapot-Chartier, M.; Goesmann, A.; Gasson, M.; Kuipers, O.; et al. Specificity and selectivity determinants of peptide transport in Lactococcus lactis and other microorganisms. Mol. Microbiol. 2005, 57, 640–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingsford, C.L.; Ayanbule, K.; Salzberg, S.L. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007, 8, R22. [Google Scholar] [CrossRef] [PubMed]
- Macke, T.J.; Ecker, D.J.; Gutell, R.R.; Gautheret, D.; Case, D.A.; Sampath, R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001, 29, 4724–4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautheret, D.; Lambert, A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol. 2001, 313, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, S.; Joshi, T.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs. RNA 2012, 18, 900–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RNA-Seq/get_gc_content.pl at Master·Spundhir/RNA-Seq·GitHub. Available online: https://github.com/spundhir/RNA-Seq/blob/master/get_gc_content.pl (accessed on 21 August 2018).
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Ackermann, H.-W.; Kropinski, A.M. Taxonomic Proposal to the ICTV Executive Committee. Available online: https://talk.ictvonline.org/ICTV/proposals/2008.011a-gB.v3.Picovirinae.pdf (accessed on 12 November 2018).
- Crucitti, P.; Lázaro, J.M.; Beneš, V.; Salas, M. Bacteriophage φ29 early protein p17 is conditionally required for the first rounds of viral DNA replication. Gene 1998, 223, 135–142. [Google Scholar] [CrossRef]
- Chung, C.-H.; Walter, M.H.; Yang, L.; Chen, S.-C.; Winston, V.; Thomas, M.A. Predicting genome terminus sequences of Bacillus cereus-group bacteriophage using next generation sequencing data. BMC Genom. 2017, 18, 350. [Google Scholar] [CrossRef] [PubMed]
- Erill, I.; Caruso, S.M. 2016 UMBC Phage Hunters Complete Genome Sequences of Three phi29-Like Bacillus cereus Group Podoviridae. Genome Announc. 2017, 5, e00701-17. [Google Scholar] [CrossRef] [PubMed]
- Redondo, R.A.F.; Kupczok, A.; Stift, G.; Bollback, J.P. Complete Genome Sequence of the Novel Phage MG-B1 Infecting Bacillus weihenstephanensis. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasko, D.A.; Altherr, M.R.; Han, C.S.; Ravel, J. Genomics of the Bacillus cereus group of organisms. FEMS Microbiol. Rev. 2005, 29, 303–329. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Cheng, B.; Zhang, S.; Wang, N.; Fang, Y.; Zhang, Q.; Kuang, A.; Lin, L.; Ji, X.; Wei, Y. Complete genome sequence of the cold-active bacteriophage VMY22 from Bacillus cereus. Virus Genes 2016, 52, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Anstead, C.A.; Korhonen, P.K.; Young, N.D.; Hall, R.S.; Jex, A.R.; Murali, S.C.; Hughes, D.S.T.; Lee, S.F.; Perry, T.; Stroehlein, A.J.; et al. Lucilia cuprina genome unlocks parasitic fly biology to underpin future interventions. Nat. Commun. 2015, 6, 7344. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Crippen, T.L.; Zheng, L.; Fields, A.T.; Yu, Z.; Ma, Q.; Wood, T.K.; Dowd, S.E.; Flores, M.; Tomberlin, J.K.; et al. A metagenomic assessment of the bacteria associated with Lucilia sericata and Lucilia cuprina (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 2015, 99, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Rodney Brister, J. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Hoppert, M.; Daniel, R.; Hertel, R. Complete Genome Sequence of vB_BveP-Goe6, a Virus Infecting Bacillus velezensis FZB42. Genome Announc. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.E. The Isolation and morphology of Some New Bacteriophages Specific for Bacillus and Acetobacter species. J. Gen. Microbiol. 1965, 41, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldentey, J.; Blanco, L.; Bamford, D.H.; Salas, M. In Vitro replication of bacteriophage PRD1 DNA. Characterization of the protein-primed initiation site. Nucleic Acids Res. 1993, 21, 3725–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Olson, N.H.; Xu, W.; Anderson, D.L.; Rossmann, M.G.; Baker, T.S.; Anderson, D.; Rossmann, M.; Baker, T.; Incardona, N.; et al. Assembly of a tailed bacterial virus and its genome release studied in three dimensions. Cell 1998, 95, 431–437. [Google Scholar] [CrossRef]
- García, J.A.; Carrascosa, J.L.; Salas, M. Assembly of the tail protein of the Bacillus subtilis phage phi 29. Virology 1983, 125, 18–30. [Google Scholar] [CrossRef]
- Gutiérrez, D.; Briers, Y.; Rodríguez-Rubio, L.; Martínez, B.; Rodríguez, A.; Lavigne, R.; García, P. Role of the Pre-neck Appendage Protein (Dpo7) from Phage vB_SepiS-phiIPLA7 as an Anti-biofilm Agent in Staphylococcal Species. Front. Microbiol. 2015, 6, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häuser, R.; Blasche, S.; Dokland, T.; Haggård-Ljungquist, E.; von Brunn, A.; Salas, M.; Casjens, S.; Molineux, I.; Uetz, P. Bacteriophage Protein-Protein Interactions; Academic Press: Cambridge, MA, USA, 2012; Volume 83, ISBN 9780123944382. [Google Scholar]
- Horcajadas, J.A.; Meijer, W.J.; Rojo, F.; Salas, M. Analysis of early promoters of the Bacillus bacteriophage GA-1. J. Bacteriol. 2001, 183, 6965–6970. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Heras, G.; Salas, M.; Bravo, A. A uracil-DNA glycosylase inhibitor encoded by a non-uracil containing viral DNA. J. Biol. Chem. 2006, 281, 7068–7074. [Google Scholar] [CrossRef] [PubMed]
- Westers, H.; Dorenbos, R.; van Dijl, J.M.; Kabel, J.; Flanagan, T.; Devine, K.M.; Jude, F.; Seror, S.J.; Beekman, A.C.; Darmon, E.; et al. Genome engineering reveals large dispensable regions in Bacillus subtilis. Mol. Biol. Evol. 2003, 20, 2076–2090. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Dietrich, S.; Hoppert, M.; Hertel, R. A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages. Viruses 2018, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Martín, A.C.; López, R.; García, P. Analysis of the complete nucleotide sequence and functional organization of the genome of Streptococcus pneumoniae bacteriophage Cp-1. J. Virol. 1996, 70, 3678–3687. [Google Scholar] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilling, T.; Hoppert, M.; Hertel, R. Genomic Analysis of the Recent Viral Isolate vB_BthP-Goe4 Reveals Increased Diversity of φ29-Like Phages. Viruses 2018, 10, 624. https://doi.org/10.3390/v10110624
Schilling T, Hoppert M, Hertel R. Genomic Analysis of the Recent Viral Isolate vB_BthP-Goe4 Reveals Increased Diversity of φ29-Like Phages. Viruses. 2018; 10(11):624. https://doi.org/10.3390/v10110624
Chicago/Turabian StyleSchilling, Tobias, Michael Hoppert, and Robert Hertel. 2018. "Genomic Analysis of the Recent Viral Isolate vB_BthP-Goe4 Reveals Increased Diversity of φ29-Like Phages" Viruses 10, no. 11: 624. https://doi.org/10.3390/v10110624
APA StyleSchilling, T., Hoppert, M., & Hertel, R. (2018). Genomic Analysis of the Recent Viral Isolate vB_BthP-Goe4 Reveals Increased Diversity of φ29-Like Phages. Viruses, 10(11), 624. https://doi.org/10.3390/v10110624