Alston Virus, a Novel Paramyxovirus Isolated from Bats Causes Upper Respiratory Tract Infection in Experimentally Challenged Ferrets

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus Isolation
2.3. Viruses
2.4. Parainfluenza Virus 5 Sequences
2.5. Sequencing
2.5.1. Whole-Genome Sequencing
2.5.2. Confirmation of Genome Termini
2.5.3. Amplicon Sequencing
2.6. Virus Quantification
2.7. Growth Kinetics Assay
2.8. Immunofluorescence Assay
2.9. Neutralisation Assay
2.10. Australian Flying Fox Serology
2.11. Animal Experiments
2.11.1. Study 1
2.11.2. Study 2
2.11.3. Analysis of Animal Infection Study Samples
2.11.4. Histology
2.12. Antibodies
2.13. Protein Prediction
2.14. Sialidase Assay
3. Results
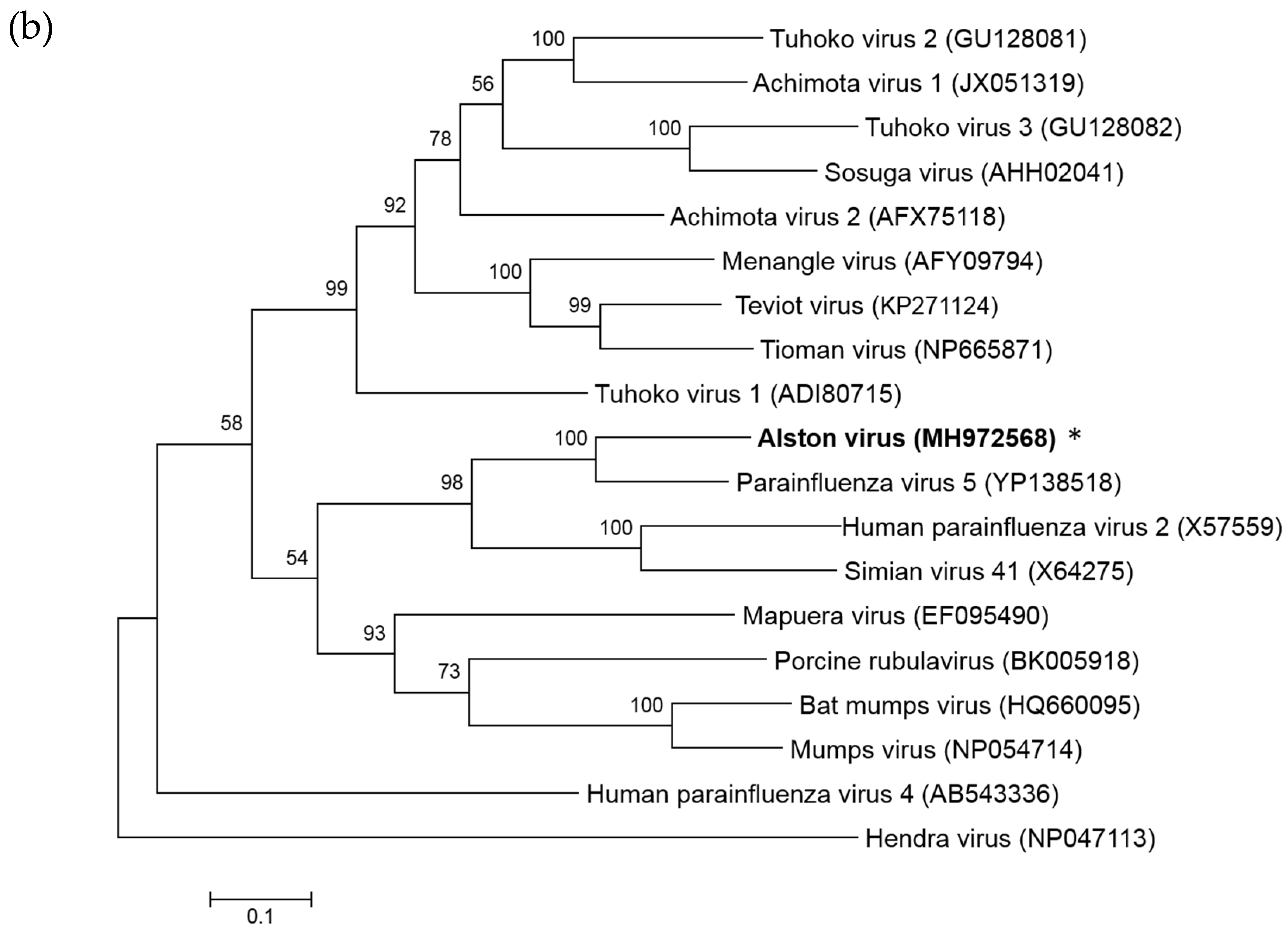
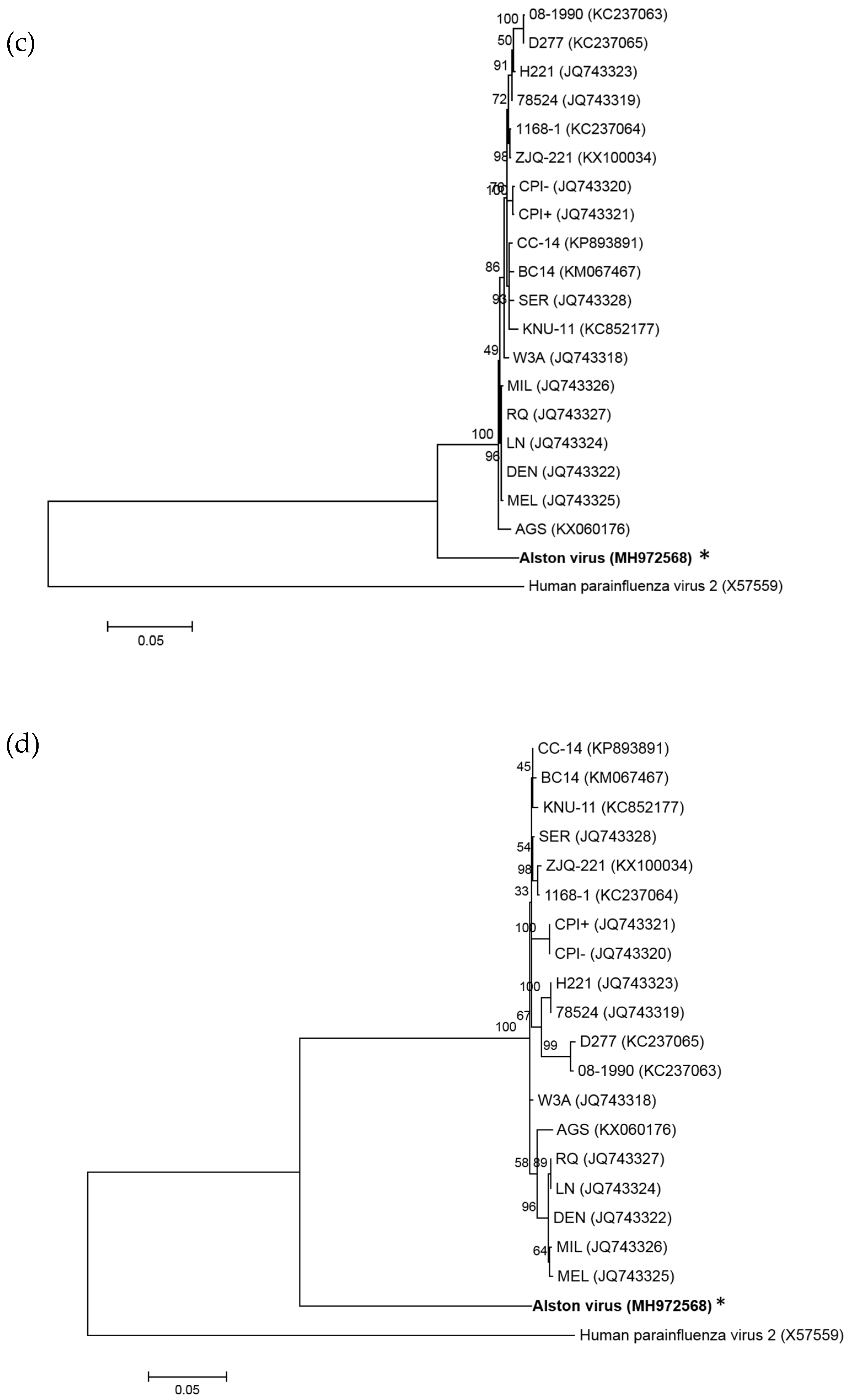
3.1. Isolation of a Novel Bat-Borne Rubulavirus
3.2. Analysis of the AlsPV Whole-Genome Sequence
3.3. Analysis of Deduced Amino Acid Sequences
3.3.1. SH
3.3.2. HN
3.3.3. RNA Editing of the P Gene
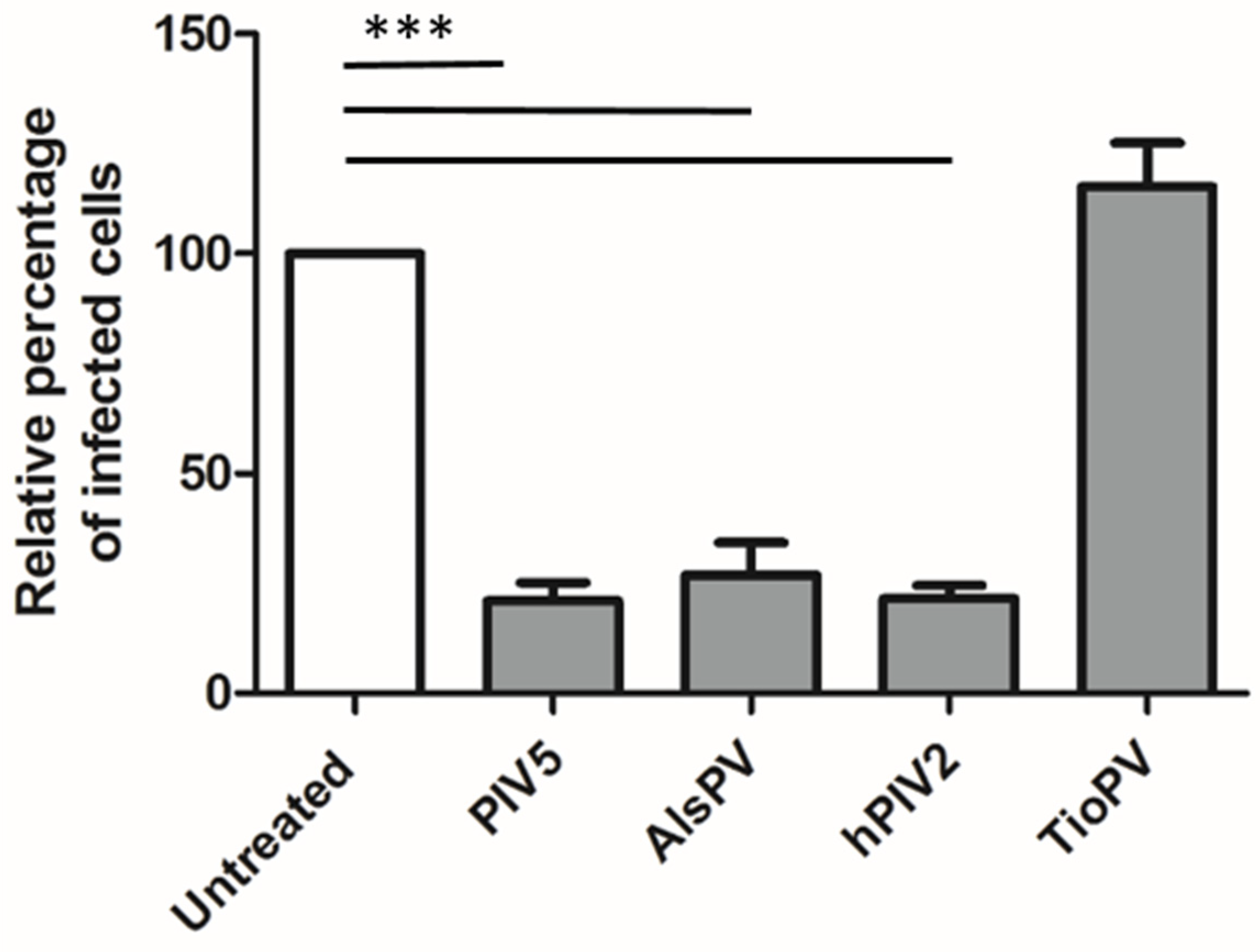
3.4. AlsPV is Antigenically Related to PIV5
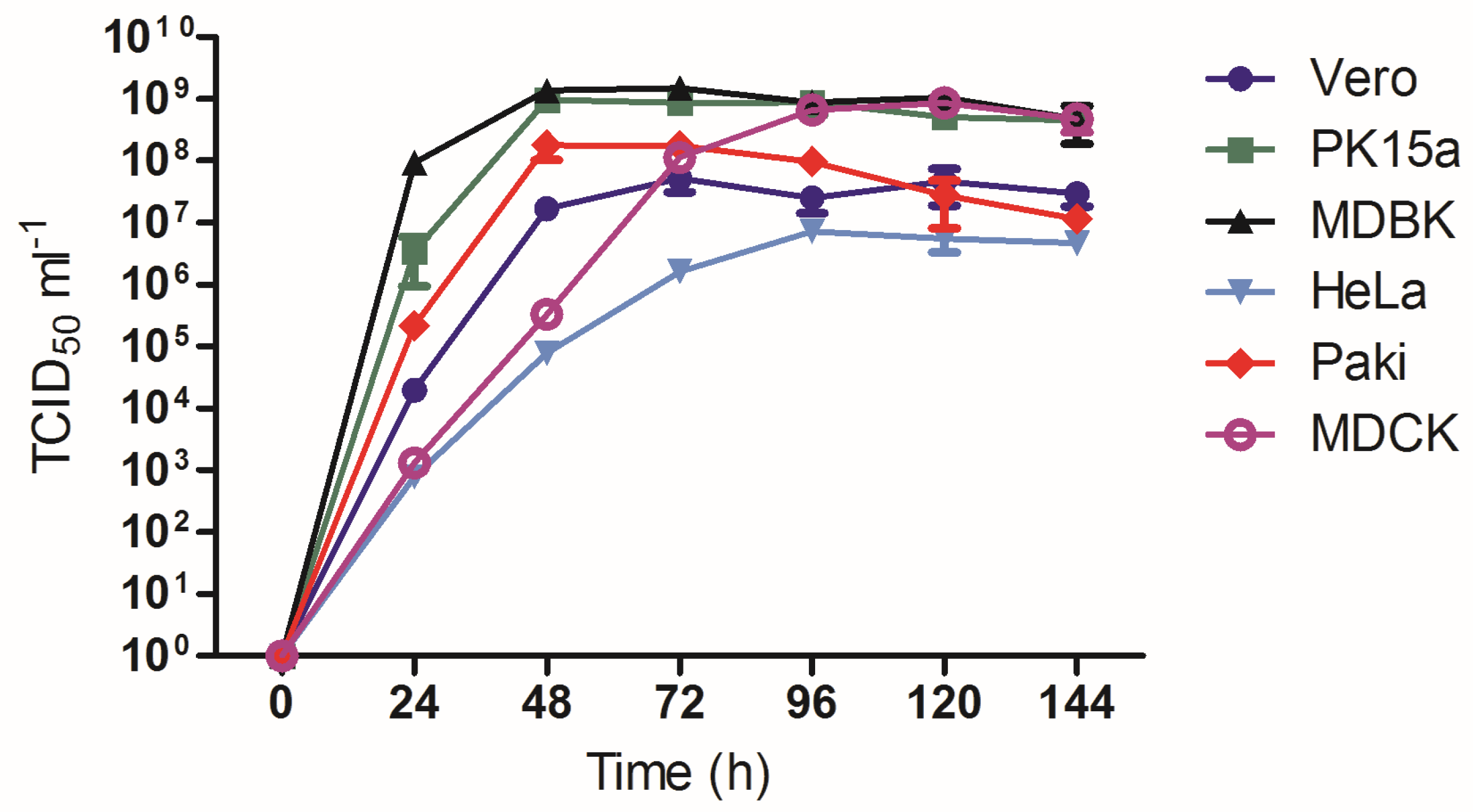
3.5. Growth Analysis of AlsPV in Mammalian Cell Lines
3.6. AlsPV Neutralising Antisera Are Prevalent in Grey Headed Flying Foxes
3.7. Animal Infection Studies
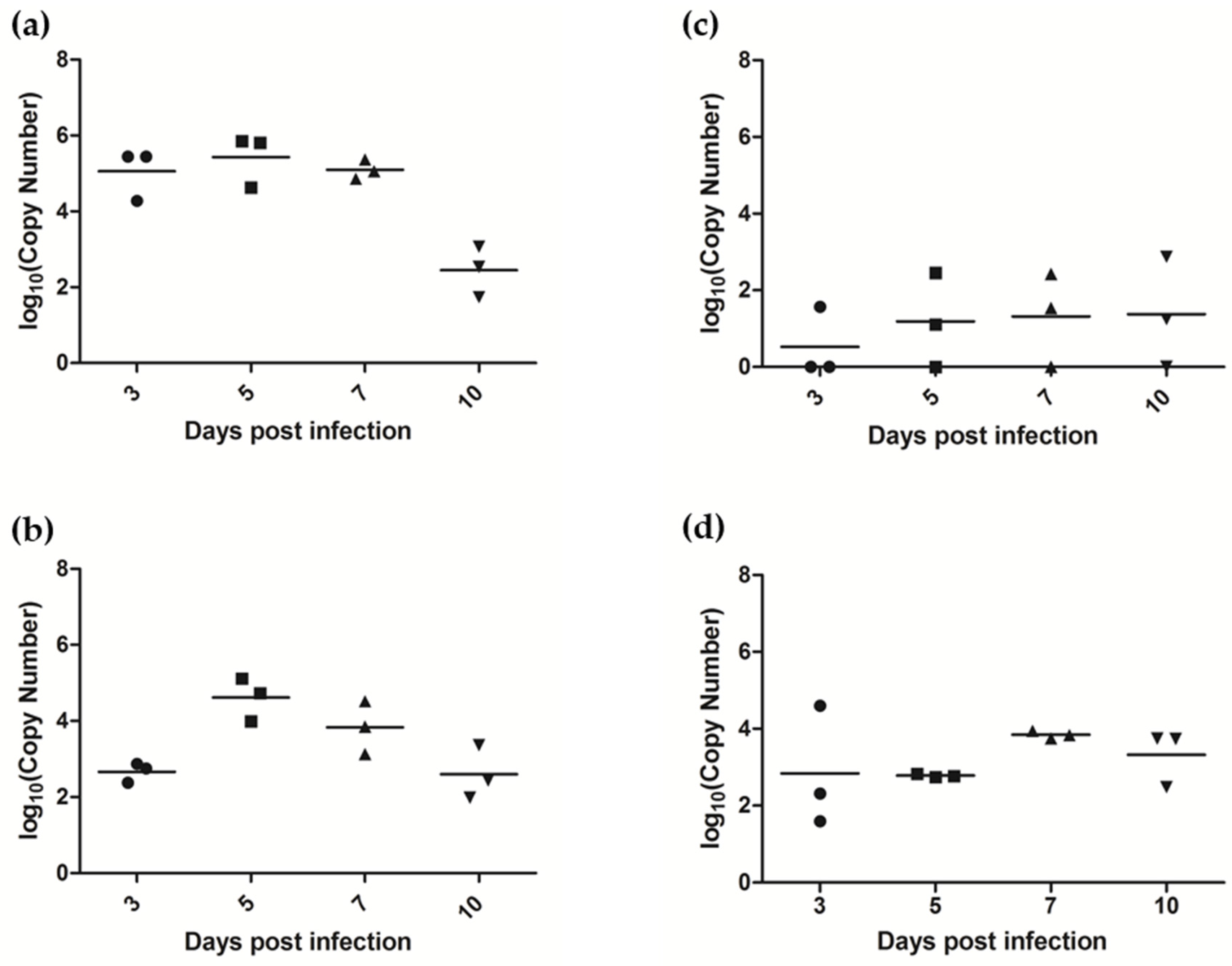
3.7.1. AlsPV Is Shed in Ferret Respiratory Secretions
3.7.2. AlsPV Infects the Ferret Upper Respiratory Tract and Olfactory Lobe of the Brain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of sars-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Field, H.; Crameri, G.; Kung, N.Y.; Wang, L.F. Ecological aspects of hendra virus. Curr. Top. Microbiol. Immunol. 2012, 359, 11–23. [Google Scholar] [PubMed]
- Chua, K.B.; Koh, C.L.; Hooi, P.S.; Wee, K.F.; Khong, J.H.; Chua, B.H.; Chan, Y.P.; Lim, M.E.; Lam, S.K. Isolation of Nipah virus from malaysian island flying-foxes. Microbes Infect. 2002, 4, 145–151. [Google Scholar] [CrossRef]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef] [PubMed]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plowright, R.K.; Foley, P.; Field, H.E.; Dobson, A.P.; Foley, J.E.; Eby, P.; Daszak, P. Urban habituation, ecological connectivity and epidemic dampening: The emergence of hendra virus from flying foxes (Pteropus spp.). Proc. Biol Sci. 2011, 278, 3703–3712. [Google Scholar] [CrossRef] [PubMed]
- Tait, J.; Perotto-Baldivieso, H.L.; McKeown, A.; Westcott, D.A. Are flying-foxes coming to town? Urbanisation of the spectacled flying-fox (Pteropus conspicillatus) in Australia. PLoS ONE 2014, 9, e109810. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, N.D.; Dunavan, C.P.; Diamond, J. Origins of major human infectious diseases. Nature 2007, 447, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Young, C.C.; Olival, K.J. Optimizing viral discovery in bats. PLoS ONE 2016, 11, e0149237. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Senior, A.M.; Di Giallonardo, F.; Holmes, E.C. Virological factors that increase the transmissibility of emerging human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 4170–4175. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2017). Archi. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Muller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, J.; Smith, C.; Smith, I.; de Jong, C.; Todd, S.; Melville, D.; Broos, A.; Crameri, S.; Haining, J.; Marsh, G.; et al. Isolation of multiple novel paramyxoviruses from pteropid bat urine. J. Gen. Virol. 2015, 96, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Collins, P.L.; Fouchier, R.A.M.; Kurath, G.; Lamb, R.A.; Randall, R.E.; Rima, B.K. Family paramyxoviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Lamb, R.A.; Parks, G.D. Paramyxoviridae. In Fields virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 957–995. [Google Scholar]
- Lin, Y.; Bright, A.C.; Rothermel, T.A.; He, B. Induction of apoptosis by paramyxovirus simian virus 5 lacking a small hydrophobic gene. J. Virol. 2003, 77, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Ford, R.B. Canine infectious respiratory disease. In Infectious Diseases of the Dog and Cat, 4th ed.; Greene, C.E., Ed.; Elsevier Health Sciences: St Louis, MO, USA, 2013; pp. 55–65. [Google Scholar]
- Hull, R.N.; Minner, J.R.; Smith, J.W. New viral agents recovered from tissue cultures of monkey kidney cells. I. Origin and properties of cytopathogenic agents s.V.1, s.V.2, s.V.4, s.V.5, s.V.6, s.V.11, s.V.12 and s.V.15. Am. J. Hyg. 1956, 63, 204–215. [Google Scholar] [PubMed]
- Chatziandreou, N.; Stock, N.; Young, D.; Andrejeva, J.; Hagmaier, K.; McGeoch, D.J.; Randall, R.E. Relationships and host range of human, canine, simian and porcine isolates of simian virus 5 (parainfluenza virus 5). J. Gen. Virol. 2004, 85, 3007–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, N.; Zhang, S.; Zhang, F.; Lian, H.; Hu, R. Parainfluenza virus 5 as possible cause of severe respiratory disease in calves, China. Emerg. Infect. Dis. 2015, 21, 2242–2244. [Google Scholar] [CrossRef] [PubMed]
- Crameri, G.; Todd, S.; Grimley, S.; McEachern, J.A.; Marsh, G.A.; Smith, C.; Tachedjian, M.; De Jong, C.; Virtue, E.R.; Yu, M.; et al. Establishment, immortalisation and characterisation of pteropid bat cell lines. PLoS ONE 2009, 4, e8266. [Google Scholar] [CrossRef] [PubMed]
- Field, H.; de Jong, C.; Melville, D.; Smith, C.; Smith, I.; Broos, A.; Kung, Y.H.; McLaughlin, A.; Zeddeman, A. Hendra virus infection dynamics in Australian fruit bats. PLoS ONE 2011, 6, e28678. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.A.; Smith, C.; Marsh, G.A.; Field, H.; Wang, L.F. Evidence of bat origin for menangle virus, a zoonotic paramyxovirus first isolated from diseased pigs. J. Gen. Virol. 2012, 93, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Chern, S.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Renner, D.W.; Albert, I.; Szpara, M.L. Viramp: A galaxy-based viral genome assembly pipeline. Gigascience 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Bestebroer, T.M.; Spronken, M.I.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Rapid sequencing of the non-coding regions of influenza a virus. J. Virol. Methods 2007, 139, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, M.; Zhang, H.; Wang, H.Y.; Wang, L.F. Improved rapid amplification of cDNA ends (RACE) for mapping both the 5′ and 3′ terminal sequences of paramyxovirus genomes. J. Virol. Methods 2005, 130, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Ameri. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Simmons, C.P.; Bernasconi, N.L.; Suguitan, A.L.; Mills, K.; Ward, J.M.; Chau, N.V.; Hien, T.T.; Sallusto, F.; Ha do, Q.; Farrar, J.; et al. Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med. 2007, 4, e178. [Google Scholar] [CrossRef] [PubMed]
- Bowden, T.R.; Bingham, J.; Harper, J.A.; Boyle, D.B. Menangle virus, a pteropid bat paramyxovirus infectious for pigs and humans, exhibits tropism for secondary lymphoid organs and intestinal epithelium in weaned pigs. J. Gen. Virol. 2012, 93, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kall, L.; Krogh, A.; Sonnhammer, E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Boyd, V.; Smith, I.; Crameri, G.; Burroughs, A.L.; Durr, P.A.; White, J.; Cowled, C.; Marsh, G.A.; Wang, L.F. Development of multiplexed bead arrays for the simultaneous detection of nucleic acid from multiple viruses in bat samples. J. Virol. Methods 2015, 223, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.J.; Boyle, D.B.; Eaton, B.T.; Wang, L.F. Full-length genome sequence of mossman virus, a novel paramyxovirus isolated from rodents in australia. Virology 2003, 317, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, S.W.; Richardson, C.D.; Lamb, R.A. Cell surface expression and orientation in membranes of the 44-amino-acid SH protein of simian virus 5. J. Virol. 1988, 62, 2347–2357. [Google Scholar] [PubMed]
- Jorgensen, E.D.; Collins, P.L.; Lomedico, P.T. Cloning and nucleotide sequence of newcastle disease virus hemagglutinin-neuraminidase mRNA: Identification of a putative sialic acid binding site. Virology 1987, 156, 12–24. [Google Scholar] [CrossRef]
- Mirza, A.M.; Deng, R.; Iorio, R.M. Site-directed mutagenesis of a conserved hexapeptide in the paramyxovirus hemagglutinin-neuraminidase glycoprotein: Effects on antigenic structure and function. J. Virol. 1994, 68, 5093–5099. [Google Scholar] [PubMed]
- Rima, B.K.; Gatherer, D.; Young, D.F.; Norsted, H.; Randall, R.E.; Davison, A.J. Stability of the parainfluenza virus 5 genome revealed by deep sequencing of strains isolated from different hosts and following passage in cell culture. J. Virol. 2014, 88, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Capraro, G.A.; Johnson, J.B.; Kock, N.D.; Parks, G.D. Virus growth and antibody responses following respiratory tract infection of ferrets and mice with WT and P/V mutants of the paramyxovirus simian virus 5. Virology 2008, 376, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Durchfeld, B.; Baumgartner, W.; Krakowka, S. Intranasal infection of ferrets (mustela putorius furo) with canine parainfluenza virus. Zentralbl. Veterinarmed. B. 1991, 38, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Durrant, D.M.; Ghosh, S.; Klein, R.S. The olfactory bulb: An immunosensory effector organ during neurotropic viral infections. ACS Chem. Neurosci. 2016, 7, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Detje, C.N.; Meyer, T.; Schmidt, H.; Kreuz, D.; Rose, J.K.; Bechmann, I.; Prinz, M.; Kalinke, U. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 2009, 182, 2297–2304. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Feature | Coding Region | Virus | Length (nt) | Length (aa) |
|---|---|---|---|---|
| 3′ leader | AlsPV | 55 | ||
| PIV5 | 55 | |||
| N | AlsPV | 1530 | 509 | |
| PIV5 | 1530 | 509 | ||
| P | V | AlsPV | 669 | 222 |
| PIV5 | 669 | 222 | ||
| P | AlsPV | 1179 | 392 | |
| PIV5 | 1179 | 392 | ||
| W | AlsPV | 516 | 171 | |
| PIV5 | 516 | 171 | ||
| M | AlsPV | 1134 | 377 | |
| PIV5 | 1134 | 377 | ||
| F | AlsPV | 1629 | 542 | |
| PIV5 | 1590 | 529 * | ||
| SH | AlsPV | 135 | 44 | |
| PIV5 | 135 | 44 | ||
| HN | AlsPV | 1698 | 565 | |
| PIV5 | 1698 | 565 | ||
| L | AlsPV | 6768 | 2255 | |
| PIV5 | 6768 | 2255 | ||
| 5′ Trailer | AlsPV | 31 | ||
| PIV5 | 31 |
| N | P | V | W | M | F | SH | HN | L | |
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | 76 | 79.4 | 80.6 | 76.9 | 76.6 | 72.5 | 63 | 71 | 76.7 |
| Amino acid | 91 | 86 | 89 | 86.6 | 93 | 85 | 61 | 81 | 92 |
| Sera from | AlsPV | PIV5 | hPIV2 | |
|---|---|---|---|---|
| Infected with | ||||
| AlsPV | 202 | 14 | <10 | |
| PIV5 | <10 | 160 | <10 | |
| hPIV2 | <10 | <10 | 226 | |
| No. Positive | Percentage Positive | |
|---|---|---|
| Pteropus sp.* | 4/59 | 6.8 |
| P. scapulatus | 0/15 | 0 |
| P. alecto | 1/26 | 3.8 |
| P. poliocephalus | 5/20 | 25 |
| Total | 10/120 | 8.3 |
| Day 7 | Day 10 | Day 14 | Day 21 | |
|---|---|---|---|---|
| Ferret 1 | <10 | <10 | 13 | 101 |
| Ferret 2 | <10 | 13 | 13 | 40 |
| Ferret 3 | <10 | <10 | 32 | 202 |
| Dpi 3 | Dpi 5 | Dpi 7 | Dpi 10 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ferret # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| Nasal turbinates | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/− | +/− | +/+ | |
| Tonsil | +/+ | +/− | +/− | +/+ | +/+ | +/+ | +/− | +/− | +/− | +/− | +/− | +/− | |
| Retropharyngeal L.N. | +/− | - | - | +/− | - | +/− | +/− | - | +/− | +/− | - | +/− | |
| Trachea | - | - | - | - | - | +/− | +/− | - | - | - | - | +/− | |
| Lung (hilus) | - | - | - | +/− | - | +/− | - | - | - | - | - | - | |
| Lung (peripheral) | - | - | - | +/− | - | - | - | - | +/− | - | - | - | |
| Bronchial L.N. | - | - | +/− | - | - | - | - | - | - | +/− | +/− | - | |
| Heart | - | - | - | - | - | +/− | - | - | - | - | - | - | |
| Liver | - | - | - | - | - | - | - | - | - | - | - | - | |
| Kidney | - | - | - | - | - | - | - | - | - | - | - | - | |
| Spleen | - | - | - | - | - | - | - | - | - | - | - | - | |
| Brain | +/+ | +/− | +/− | +/− | +/− | +/− | +/− | +/− | +/+ | +/− | +/− | +/− | |
| Small intestine | - | - | - | - | - | +/− | - | - | - | - | - | - | |
| Large intestine | - | - | - | - | - | - | - | - | - | - | - | - | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, R.I.; Tachedjian, M.; Rowe, B.; Clayton, B.A.; Layton, R.; Bergfeld, J.; Wang, L.-F.; Smith, I.L.; Marsh, G.A. Alston Virus, a Novel Paramyxovirus Isolated from Bats Causes Upper Respiratory Tract Infection in Experimentally Challenged Ferrets. Viruses 2018, 10, 675. https://doi.org/10.3390/v10120675
Johnson RI, Tachedjian M, Rowe B, Clayton BA, Layton R, Bergfeld J, Wang L-F, Smith IL, Marsh GA. Alston Virus, a Novel Paramyxovirus Isolated from Bats Causes Upper Respiratory Tract Infection in Experimentally Challenged Ferrets. Viruses. 2018; 10(12):675. https://doi.org/10.3390/v10120675
Chicago/Turabian StyleJohnson, Rebecca I., Mary Tachedjian, Brenton Rowe, Bronwyn A. Clayton, Rachel Layton, Jemma Bergfeld, Lin-Fa Wang, Ina L. Smith, and Glenn A. Marsh. 2018. "Alston Virus, a Novel Paramyxovirus Isolated from Bats Causes Upper Respiratory Tract Infection in Experimentally Challenged Ferrets" Viruses 10, no. 12: 675. https://doi.org/10.3390/v10120675
APA StyleJohnson, R. I., Tachedjian, M., Rowe, B., Clayton, B. A., Layton, R., Bergfeld, J., Wang, L. -F., Smith, I. L., & Marsh, G. A. (2018). Alston Virus, a Novel Paramyxovirus Isolated from Bats Causes Upper Respiratory Tract Infection in Experimentally Challenged Ferrets. Viruses, 10(12), 675. https://doi.org/10.3390/v10120675