Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs


{kind=link}
Abstract
:1. Introduction
2. “Shock and Kill” Method
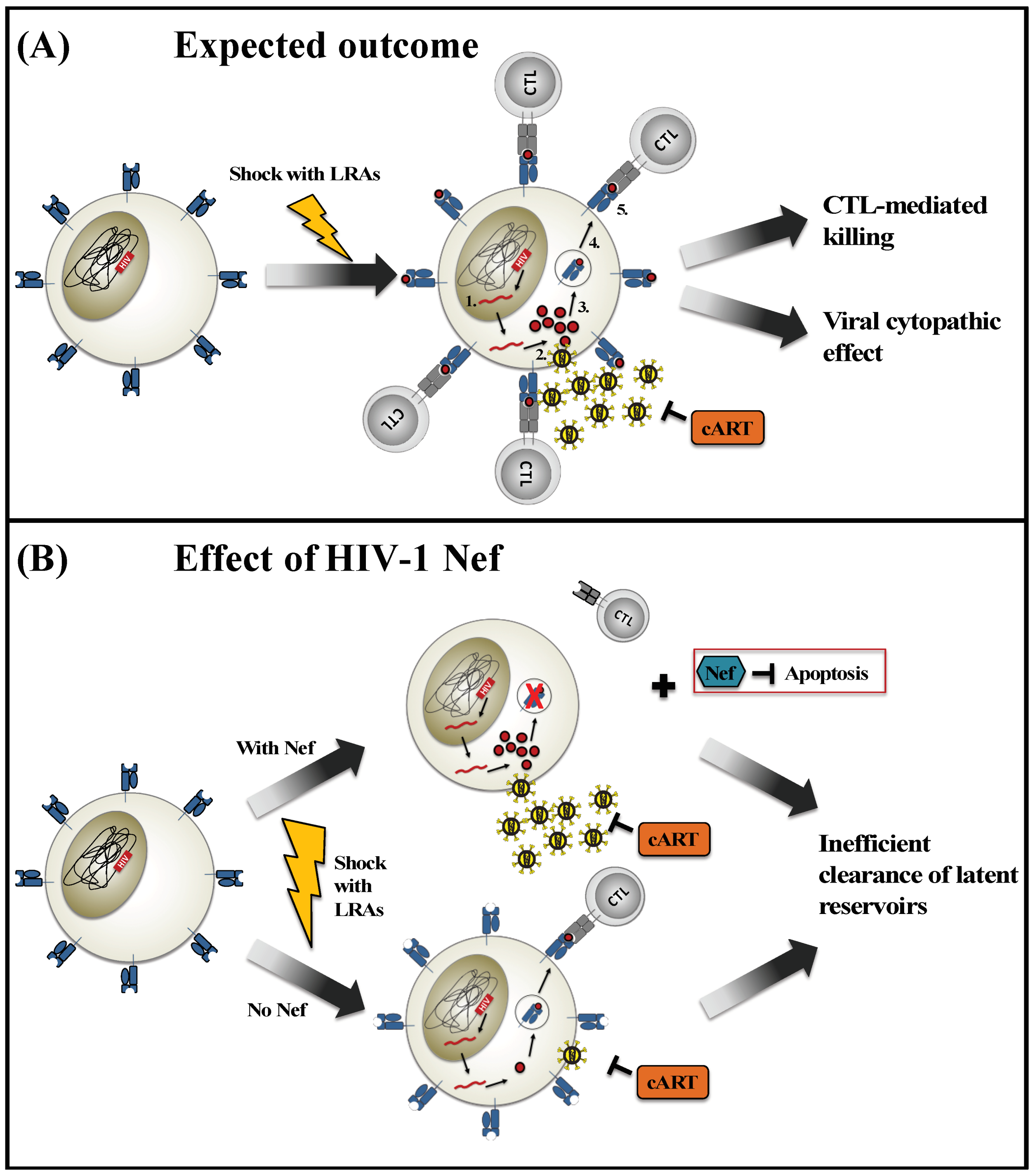
2.1. Inefficient Viral Reactivation Using LRAs
2.2. Ineffective Clearance of Reactivated Cells
3. Modulation of HIV-Infected Cells by Nef
4. The Double-Edged Effect of HIV-1 Nef
4.1. How Nef Might Enhance “Shock and Kill” Strategies
4.2. How Nef Might Impair “Shock and Kill” Strategies
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hutter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the Berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS/WHO. Global HIV & AIDS Statistics—2018 Fact Sheet; UNAIDS/WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 2014, 161, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Delagreverie, H.M.; Delaugerre, C.; Lewin, S.R.; Deeks, S.G.; Li, J.Z. Ongoing Clinical Trials of Human Immunodeficiency Virus Latency-Reversing and Immunomodulatory Agents. Open Forum Infect. Dis. 2016, 3, ofw189. [Google Scholar] [CrossRef] [PubMed]
- Thorlund, K.; Horwitz, M.S.; Fife, B.T.; Lester, R.; Cameron, D.W. Landscape review of current HIV ‘kick and kill’ cure research—Some kicking, not enough killing. BMC Infect. Dis. 2017, 17, 595. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korin, Y.D.; Brooks, D.G.; Brown, S.; Korotzer, A.; Zack, J.A. Effects of prostratin on T-cell activation and human immunodeficiency virus latency. J. Virol. 2002, 76, 8118–8123. [Google Scholar] [CrossRef] [PubMed]
- Mehla, R.; Bivalkar-Mehla, S.; Zhang, R.; Handy, I.; Albrecht, H.; Giri, S.; Nagarkatti, P.; Nagarkatti, M.; Chauhan, A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS ONE 2010, 5, e11160. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals with Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Sereti, I.; Dunham, R.M.; Spritzler, J.; Aga, E.; Proschan, M.A.; Medvik, K.; Battaglia, C.A.; Landay, A.L.; Pahwa, S.; Fischl, M.A.; et al. IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood 2009, 113, 6304–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Stellbrink, H.J.; van Lunzen, J.; Westby, M.; O’Sullivan, E.; Schneider, C.; Adam, A.; Weitner, L.; Kuhlmann, B.; Hoffmann, C.; Fenske, S.; et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial). AIDS 2002, 16, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; Blankson, J.N. The Effect of Latency Reversal Agents on Primary CD8+ T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clutton, G.; Xu, Y.; Baldoni, P.L.; Mollan, K.R.; Kirchherr, J.; Newhard, W.; Cox, K.; Kuruc, J.D.; Kashuba, A.; Barnard, R.; et al. The differential short- and long-term effects of HIV-1 latency-reversing agents on T cell function. Sci. Rep. 2016, 6, 30749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Byrn, R.; Groopman, J.; Baltimore, D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: Evidence for differential gene expression. J. Virol. 1989, 63, 3708–3713. [Google Scholar] [PubMed]
- Robert-Guroff, M.; Popovic, M.; Gartner, S.; Markham, P.; Gallo, R.C.; Reitz, M.S. Structure and expression of tat-, rev-, and nef-specific transcripts of human immunodeficiency virus type 1 in infected lymphocytes and macrophages. J. Virol. 1990, 64, 3391–3398. [Google Scholar] [PubMed]
- Klotman, M.E.; Kim, S.; Buchbinder, A.; DeRossi, A.; Baltimore, D.; Wong-Staal, F. Kinetics of expression of multiply spliced RNA in early human immunodeficiency virus type 1 infection of lymphocytes and monocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 5011–5015. [Google Scholar] [CrossRef] [PubMed]
- Zaunders, J.J.; Geczy, A.F.; Dyer, W.B.; McIntyre, L.B.; Cooley, M.A.; Ashton, L.J.; Raynes-Greenow, C.H.; Learmont, J.; Cooper, D.A.; Sullivan, J.S. Effect of long-term infection with nef-defective attenuated HIV type 1 on CD4+ and CD8+ T lymphocytes: Increased CD45RO+CD4+ T lymphocytes and limited activation of CD8+ T lymphocytes. AIDS Res. Hum. Retroviruses 1999, 15, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.N.; Dikeakos, J.D. HIV-1 Nef: A master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 2015, 1850, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making Sense of Multifunctional Proteins: Human Immunodeficiency Virus Type 1 Accessory and Regulatory Proteins and Connections to Transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.V.; Miller, A.D. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 1991, 350, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.; Konner, J.; Landau, N.R.; Lenburg, M.E.; Trono, D. Nef induces CD4 endocytosis: Requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 1994, 76, 853–864. [Google Scholar] [CrossRef]
- Laguette, N.; Bregnard, C.; Bouchet, J.; Benmerah, A.; Benichou, S.; Basmaciogullari, S. Nef-induced CD4 endocytosis in human immunodeficiency virus type 1 host cells: Role of p56lck kinase. J. Virol. 2009, 83, 7117–7128. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Chang, S.H.; Kwon, J.H.; Rhee, S.S. HIV-1 Nef plays an essential role in two independent processes in CD4 down-regulation: Dissociation of the CD4-p56(lck) complex and targeting of CD4 to lysosomes. Virology 1999, 257, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.J.; Janvier, K.; Chandrasekhar, S.; Sekaly, R.P.; Bonifacino, J.S.; Venkatesan, S. CD4 down-regulation by HIV-1 and simian immunodeficiency virus (SIV) Nef proteins involves both internalization and intracellular retention mechanisms. J. Biol. Chem. 2005, 280, 7413–7426. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Oran, A.E.; Cullen, B.R. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol. CB 1999, 9, 613–621. [Google Scholar] [CrossRef]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef]
- Arganaraz, E.R.; Schindler, M.; Kirchhoff, F.; Cortes, M.J.; Lama, J. Enhanced CD4 down-modulation by late stage HIV-1 nef alleles is associated with increased Env incorporation and viral replication. J. Biol. Chem. 2003, 278, 33912–33919. [Google Scholar] [CrossRef] [PubMed]
- Wildum, S.; Schindler, M.; Munch, J.; Kirchhoff, F. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Desormeaux, A.; Medjahed, H.; Gharsallah, N.E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, B.H.; et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Prevost, J.; Richard, J.; Medjahed, H.; Alexander, A.; Jones, J.; Kappes, J.C.; Ochsenbauer, C.; Finzi, A. Incomplete Downregulation of CD4 Expression Affects HIV-1 Env Conformation and Antibody-Dependent Cellular Cytotoxicity Responses. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Alsahafi, N.; Ding, S.; Richard, J.; Markle, T.; Brassard, N.; Walker, B.; Lewis, G.K.; Kaufmann, D.E.; Brockman, M.A.; Finzi, A. Nef Proteins from HIV-1 Elite Controllers Are Inefficient at Preventing Antibody-Dependent Cellular Cytotoxicity. J. Virol. 2015, 90, 2993–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Mangasarian, A.; Piguet, V.; Wang, J.K.; Chen, Y.L.; Trono, D. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 1999, 73, 1964–1973. [Google Scholar] [PubMed]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Safrit, J.T.; Cao, Y.; Andrews, C.A.; McLeod, G.; Borkowsky, W.; Farthing, C.; Ho, D.D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994, 68, 4650–4655. [Google Scholar] [PubMed]
- Borrow, P.; Lewicki, H.; Wei, X.; Horwitz, M.S.; Peffer, N.; Meyers, H.; Nelson, J.A.; Gairin, J.E.; Hahn, B.H.; Oldstone, M.B.; et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997, 3, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Leslie, A.J.; Honeyborne, I.; Ramduth, D.; Thobakgale, C.; Chetty, S.; Rathnavalu, P.; Moore, C.; Pfafferott, K.J.; Hilton, L.; et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 2004, 432, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Altfeld, M.; Kalife, E.T.; Qi, Y.; Streeck, H.; Lichterfeld, M.; Johnston, M.N.; Burgett, N.; Swartz, M.E.; Yang, A.; Alter, G.; et al. HLA Alleles Associated with Delayed Progression to AIDS Contribute Strongly to the Initial CD8(+) T Cell Response against HIV-1. PLoS Med. 2006, 3, e403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; de Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautz, B.; Wiedemann, H.; Luchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Krausslich, H.G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Biol. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, T.; Yang, J.; Lin, Y.; Shi, J.; Zhang, X.; Frabutt, D.A.; Zeng, X.; Li, S.; Venta, P.J.; et al. Identification of SERINC5-001 as the Predominant Spliced Isoform for HIV-1 Restriction. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautz, B.; Pierini, V.; Wombacher, R.; Stolp, B.; Chase, A.J.; Pizzato, M.; Fackler, O.T. The Antagonism of HIV-1 Nef to SERINC5 Particle Infectivity Restriction Involves the Counteraction of Virion-Associated Pools of the Restriction Factor. J. Virol. 2016, 90, 10915–10927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swigut, T.; Shohdy, N.; Skowronski, J. Mechanism for down-regulation of CD28 by Nef. EMBO J. 2001, 20, 1593–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoulouze, M.I.; Sol-Foulon, N.; Blanchet, F.; Dautry-Varsat, A.; Schwartz, O.; Alcover, A. Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity 2006, 24, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.M.; Eickel, N.; Haller, C.; Schindler, M.; Fackler, O.T. Inhibition of T-cell receptor-induced actin remodeling and relocalization of Lck are evolutionarily conserved activities of lentiviral Nef proteins. J. Virol. 2009, 83, 11528–11539. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Rudolph, J.M.; Abraham, L.; Habermann, A.; Haller, C.; Krijnse-Locker, J.; Fackler, O.T. HIV-1 Nef compensates for disorganization of the immunological synapse by inducing trans-Golgi network-associated Lck signaling. Blood 2012, 119, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Rauch, S.; Fackler, O.T. HIV-1 Nef employs two distinct mechanisms to modulate Lck subcellular localization and TCR induced actin remodeling. PLoS ONE 2007, 2, e1212. [Google Scholar] [CrossRef] [PubMed]
- Witte, V.; Laffert, B.; Gintschel, P.; Krautkramer, E.; Blume, K.; Fackler, O.T.; Baur, A.S. Induction of HIV transcription by Nef involves Lck activation and protein kinase C theta raft recruitment leading to activation of ERK1/2 but not NFκB. J. Immunol. 2008, 181, 8425–8432. [Google Scholar] [CrossRef] [PubMed]
- Manninen, A.; Renkema, G.H.; Saksela, K. Synergistic activation of NFAT by HIV-1 nef and the Ras/MAPK pathway. J. Biol. Chem. 2000, 275, 16513–16517. [Google Scholar] [CrossRef] [PubMed]
- Manninen, A.; Saksela, K. HIV-1 Nef interacts with inositol trisphosphate receptor to activate calcium signaling in T cells. J. Exp. Med. 2002, 195, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Geleziunas, R.; Xu, W.; Takeda, K.; Ichijo, H.; Greene, W.C. HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 2001, 410, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Greenway, A.L.; McPhee, D.A.; Allen, K.; Johnstone, R.; Holloway, G.; Mills, J.; Azad, A.; Sankovich, S.; Lambert, P. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J. Virol. 2002, 76, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.N.; Laffert, B.; Screaton, G.R.; Kraft, M.; Wolf, D.; Kolanus, W.; Mongkolsapay, J.; McMichael, A.J.; Baur, A.S. Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor zeta chain. J. Exp. Med. 1999, 189, 1489–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthumani, K.; Choo, A.Y.; Hwang, D.S.; Premkumar, A.; Dayes, N.S.; Harris, C.; Green, D.R.; Wadsworth, S.A.; Siekierka, J.J.; Weiner, D.B. HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood 2005, 106, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, Z.; Weng, X.; Kay, D.G.; Poudrier, J.; Lowell, C.; Jolicoeur, P. The pathogenicity of human immunodeficiency virus (HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-binding domain, and disease development is delayed in the absence of Hck. J. Virol. 2001, 75, 9378–9392. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Rappaport, J. HIV cure strategists: Ignore the central nervous system at your patients’ peril. AIDS 2017, 31, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Gama, L.; Abreu, C.; Shirk, E.N.; Queen, S.E.; Beck, S.E.; Metcalf Pate, K.A.; Bullock, B.T.; Zink, M.C.; Mankowski, J.L.; Clements, J.E. SIV Latency in Macrophages in the CNS. Curr. Top. Microbiol. Immunol. 2018. [Google Scholar] [CrossRef]
- Fackler, O.T.; Luo, W.; Geyer, M.; Alberts, A.S.; Peterlin, B.M. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol. Cell 1999, 3, 729–739. [Google Scholar] [CrossRef]
- Rauch, S.; Pulkkinen, K.; Saksela, K.; Fackler, O.T. Human immunodeficiency virus type 1 Nef recruits the guanine exchange factor Vav1 via an unexpected interface into plasma membrane microdomains for association with p21-activated kinase 2 activity. J. Virol. 2008, 82, 2918–2929. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Reichman-Fried, M.; Abraham, L.; Pan, X.; Giese, S.I.; Hannemann, S.; Goulimari, P.; Raz, E.; Grosse, R.; Fackler, O.T. HIV-1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe 2009, 6, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Abraham, L.; Rudolph, J.M.; Fackler, O.T. Lentiviral Nef proteins utilize PAK2-mediated deregulation of cofilin as a general strategy to interfere with actin remodeling. J. Virol. 2010, 84, 3935–3948. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Alcover, A.; Schwartz, O. Modulation of the immunological synapse: A key to HIV-1 pathogenesis? Nat. Rev. Immunol. 2007, 7, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Zhong, Q.; Nakaya, T.; Kameoka, M.; Meguro, T.; Yamada, K.; Ikuta, K. Extracellular Nef protein regulates productive HIV-1 infection from latency. J. Immunol. 1995, 155, 5289–5298. [Google Scholar] [PubMed]
- Tobiume, M.; Fujinaga, K.; Suzuki, S.; Komoto, S.; Mukai, T.; Ikuta, K. Extracellular Nef protein activates signal transduction pathway from Ras to mitogen-activated protein kinase cascades that leads to activation of human immunodeficiency virus from latency. AIDS Res. Hum. Retroviruses 2002, 18, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Manna, S.K.; Quivy, V.; Decrion, A.Z.; Van Lint, C.; Herbein, G.; Aggarwal, B.B. Exogenous Nef protein activates NF-κB, AP-1, and c-Jun N-terminal kinase and stimulates HIV transcription in promonocytic cells. Role in AIDS pathogenesis. J. Biol. Chem. 2003, 278, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Colin, L.; Khan, K.A.; Bouchat, S.; Varin, A.; Larbi, A.; Gatot, J.S.; Kabeya, K.; Vanhulle, C.; et al. Tuning of AKT-pathway by Nef and its blockade by protease inhibitors results in limited recovery in latently HIV infected T-cell line. Sci. Rep. 2016, 6, 24090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, Y.; Otake, K.; Tashiro, M.; Adachi, A. Soluble Nef antigen of HIV-1 is cytotoxic for human CD4+ T cells. FEBS Lett. 1996, 393, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Federico, M.; Percario, Z.; Olivetta, E.; Fiorucci, G.; Muratori, C.; Micheli, A.; Romeo, G.; Affabris, E. HIV-1 Nef activates STAT1 in human monocytes/macrophages through the release of soluble factors. Blood 2001, 98, 2752–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.D.; Campbell-Sims, T.C.; Khan, M.; Lang, M.; Huang, M.B.; Bond, V.C.; Powell, M.D. HIV Type 1 Nef is released from infected cells in CD45(+) microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res. Hum. Retroviruses 2011, 27, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Takei, R.; Tashiro, M. Nef protein of HIV-1 induces apoptotic cytolysis of murine lymphoid cells independently of CD95 (Fas) and its suppression by serine/threonine protein kinase inhibitors. FEBS Lett. 1997, 417, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Alessandrini, L.; Santarcangelo, A.C.; Olivetta, E.; Ferrantelli, F.; d’Aloja, P.; Pugliese, K.; Pelosi, E.; Chelucci, C.; Mattia, G.; Peschle, C.; et al. T-tropic human immunodeficiency virus (HIV) type 1 Nef protein enters human monocyte-macrophages and induces resistance to HIV replication: A possible mechanism of HIV T-tropic emergence in AIDS. J. Gen. Virol. 2000, 81, 2905–2917. [Google Scholar] [CrossRef] [PubMed]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wittki, S.; Brau, T.; Dreyer, F.S.; Kratzel, K.; Dindorf, J.; Johnston, I.C.; Gross, S.; Kremmer, E.; Zeidler, R.; et al. HIV Nef, paxillin, and Pak1/2 regulate activation and secretion of TACE/ADAM10 proteases. Mol. Cell 2013, 49, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Muratori, C.; Cavallin, L.E.; Kratzel, K.; Tinari, A.; De Milito, A.; Fais, S.; D’Aloja, P.; Federico, M.; Vullo, V.; Fomina, A.; et al. Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 2009, 6, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Ren, Y.; Thomas, A.S.; Chan, D.; Mueller, S.; Ward, A.R.; Patel, S.; Bollard, C.M.; Cruz, C.R.; Karandish, S.; et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J. Clin. Investig. 2018, 128, 876–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujib, S.; Saiyed, A.; Fadel, S.; Bozorgzad, A.; Aidarus, N.; Yue, F.Y.; Benko, E.; Kovacs, C.; Emert-Sedlak, L.A.; Smithgall, T.E.; et al. Pharmacologic HIV-1 Nef blockade promotes CD8 T cell-mediated elimination of latently HIV-1-infected cells in vitro. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.Y.; Nishitoh, H.; Yang, X.; Ichijo, H.; Baltimore, D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 1998, 281, 1860–1863. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.; Eder, A.; Mao, M.; Bast, R.C., Jr.; Boyd, D.; Mills, G.B. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 1999, 18, 6635–6640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douek, D.C.; Brenchley, J.M.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Okamoto, Y.; Casazza, J.P.; Kuruppu, J.; Kunstman, K.; Wolinsky, S.; et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 2002, 417, 95–98. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuang, X.T.; Brockman, M.A. Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs. Viruses 2018, 10, 677. https://doi.org/10.3390/v10120677
Kuang XT, Brockman MA. Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs. Viruses. 2018; 10(12):677. https://doi.org/10.3390/v10120677
Chicago/Turabian StyleKuang, Xiaomei T., and Mark A. Brockman. 2018. "Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs" Viruses 10, no. 12: 677. https://doi.org/10.3390/v10120677
APA StyleKuang, X. T., & Brockman, M. A. (2018). Implications of HIV-1 Nef for “Shock and Kill” Strategies to Eliminate Latent Viral Reservoirs. Viruses, 10(12), 677. https://doi.org/10.3390/v10120677