Chaperoning the Mononegavirales: Current Knowledge and Future Directions


Abstract
:1. The Mononegavirales
2. Protein Folding, a Common Challenge for Both Viruses and Their Hosts
3. The Human Chaperome
3.1. The HSPA Chaperone Family
3.2. The HSPB Chaperone Family
3.3. The HSPC Chaperone Family
3.4. The Chaperonin CCT
3.5. Folding in the Endoplasmic Reticulum (ER)
4. Chaperone–Mononegavirales Interactions
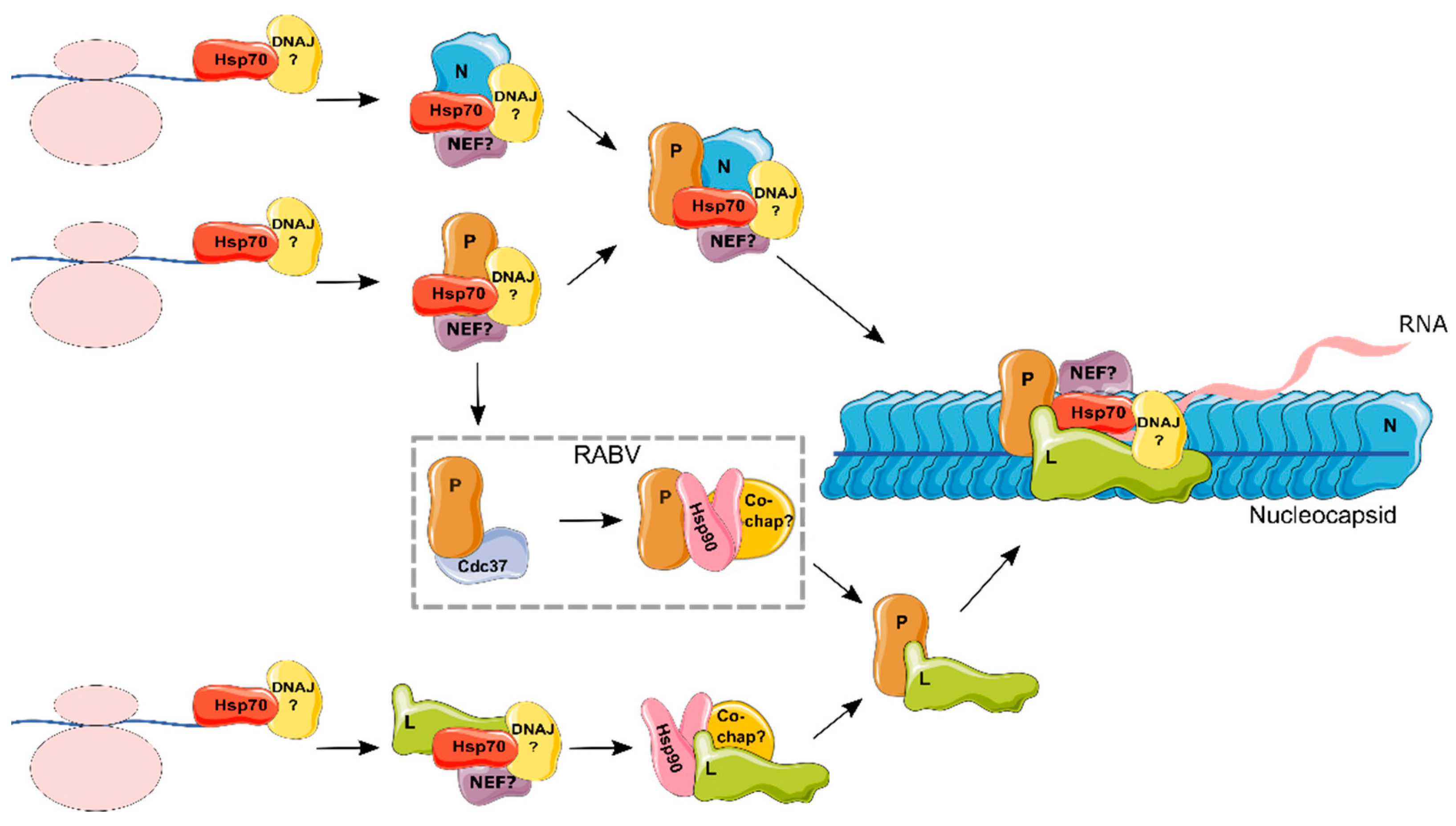
4.1. Hsp70s in the Life Cycle of the Mononegavirales
4.2. Hsp90s in the Life Cycle of the Mononegavirales
4.3. The Chaperonin CCT in the Life Cycle of the Mononegavirales
4.4. Folding the Glycoproteins of the Mononegavirales in the ER
5. Chaperone Inhibitors as Antivirals
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Amarasinghe, G.K.; Aréchiga Ceballos, N.G.; Banyard, A.C.; Basler, C.F.; Bavari, S.; Bennett, A.J.; Blasdell, K.R.; Briese, T.; Bukreyev, A.; Caì, Y.; et al. Taxonomy of the order Mononegavirales: Update 2018. Arch. Virol. 2018, 163, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Thibault, P.A.; Watkinson, R.E.; Moreira-Soto, A.; Drexler, J.F.; Lee, B. Zoonotic Potential of Emerging Paramyxoviruses: Knowns and Unknowns. Adv. Virus Res. 2017, 98, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Coltart, C.E.M.; Lindsey, B.; Ghinai, I.; Johnson, A.M.; Heymann, D.L. The Ebola outbreak, 2013-2016: Old lessons for new epidemics. Phil. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.P.J.; Barr, J.N.; Wertz, G.W. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 2004, 283, 61–119. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A. Mononegavirales. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 880–884. ISBN 9781451105636. [Google Scholar]
- Longhi, S. Nucleocapsid structure and function. Curr. Top. Microbiol. Immunol. 2009, 329, 103–128. [Google Scholar] [PubMed]
- Morin, B.; Kranzusch, P.J.; Rahmeh, A.A.; Whelan, S.P.J. The polymerase of negative-stranded RNA viruses. Curr. Opin. Virol. 2013, 3, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longhi, S.; Bloyet, L.-M.; Gianni, S.; Gerlier, D. How order and disorder within paramyxoviral nucleoproteins and phosphoproteins orchestrate the molecular interplay of transcription and replication. Cell. Mol. Life Sci. 2017, 74, 3091–3118. [Google Scholar] [CrossRef] [PubMed]
- Liljeroos, L.; Butcher, S.J. Matrix proteins as centralized organizers of negative-sense RNA virions. Front. Biosci. 2013, 18, 696–715. [Google Scholar] [CrossRef]
- Cantoni, D.; Rossman, J.S. Ebolaviruses: New roles for old proteins. PLoS Negl. Trop. Dis. 2018, 12, e0006349. [Google Scholar] [CrossRef] [PubMed]
- Komarova, A.V.; Combredet, C.; Meyniel-Schicklin, L.; Chapelle, M.; Caignard, G.; Camadro, J.-M.; Lotteau, V.; Vidalain, P.-O.; Tangy, F. Proteomic analysis of virus-host interactions in an infectious context using recombinant viruses. Mol. Cell. Proteom. 2011, 10, M110–007443. [Google Scholar] [CrossRef] [PubMed]
- de Chassey, B.; Meyniel-Schicklin, L.; Vonderscher, J.; André, P.; Lotteau, V. Virus-host interactomics: New insights and opportunities for antiviral drug discovery. Genome Med. 2014, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Eliscovich, C.; Yoon, Y.J.; Singer, R.H. Translation dynamics of single mRNAs in live cells and neurons. Science 2016, 352, 1430–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Schwanhüusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Finka, A.; Goloubinoff, P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 2013, 18, 591–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuriki, N.; Oldfield, C.J.; Uversky, V.N.; Berezovsky, I.N.; Tawfik, D.S. Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 2009, 34, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 325–355. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Dillin, A. The UPRER: Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting heat shock proteins in cancer: A promising therapeutic approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Radons, J. The Hsp90 Chaperone Machinery: An Important Hub in Protein Interaction Networks. Br. J. Med. Med. Res. 2016, 14, 1–32. [Google Scholar] [CrossRef]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Sousa, R.; Lafer, E.M. The role of molecular chaperones in clathrin mediated vesicular trafficking. Front. Mol. Biosci. 2015, 2, 26. [Google Scholar] [CrossRef]
- Craig, E.A. Hsp70 at the membrane: Driving protein translocation. BMC Biol. 2018, 16, 11. [Google Scholar] [CrossRef]
- Clerico, E.M.; Tilitsky, J.M.; Meng, W.; Gierasch, L.M. How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J. Mol. Biol. 2015, 427, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracher, A.; Verghese, J. GrpE, Hsp110/Grp170, HspBP1/Sil1 and BAG domain proteins: Nucleotide exchange factors for Hsp70 molecular chaperones. Subcell. Biochem. 2015, 78, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.K.; Ratajczak, T. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones 2011, 16, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Stürner, E.; Behl, C. The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front. Mol. Neurosci. 2017, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; de Boer, R.; Beerstra, N. The Multicolored World of the Human HSPB Family. In The Big Book on Small Heat Shock Proteins; Tanguay, R.M., Hightower, L.E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–26. ISBN 978-3-319-16077-1. [Google Scholar]
- Haslbeck, M.; Vierling, E. A first line of stress defense: Small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 2015, 427, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Morán Luengo, T.; Kityk, R.; Mayer, M.P.; Rüdiger, S.G.D. Hsp90 Breaks the Deadlock of the Hsp70 Chaperone System. Mol. Cell 2018, 70, 545–552. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, F.; Wang, R.; Li, F.; Wu, Y.; Kitazato, K.; Wang, Y. HSP90: A promising broad-spectrum antiviral drug target. Arch. Virol. 2017, 162, 3269–3282. [Google Scholar] [CrossRef]
- Geller, R.; Taguwa, S.; Frydman, J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 2012, 1823, 698–706. [Google Scholar] [CrossRef]
- Yuno, A.; Lee, M.-J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. In Chaperones: Methods and Protocols; Calderwood, S.K., Prince, T.L., Eds.; Springer: New York, NY, USA, 2018; pp. 423–441. ISBN 978-1-4939-7477-1. [Google Scholar]
- Aragonès, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS Pathog. 2010, 6, e1000797. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Dalton, K.; Frydman, J. The Mechanism and Function of Group II Chaperonins. J. Mol. Biol. 2015, 427, 2919–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yam, A.Y.; Xia, Y.; Lin, H.-T.J.; Burlingame, A.; Gerstein, M.; Frydman, J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 2008, 15, 1255–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Ploegh, H.L. Quality and quantity control at the endoplasmic reticulum. Curr. Opin. Cell Biol. 2010, 22, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Nagata, K. SUP: Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2012, 4, a015438. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Maattanen, P.; Denisov, A.Y.; Scarffe, L.; Schade, B.; Balghi, H.; Dejgaard, K.; Chen, L.Y.; Muller, W.J.; Gehring, K.; et al. An Interaction Map of Endoplasmic Reticulum Chaperones and Foldases. Mol. Cell. Proteom. 2012, 11, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Kubota, T.; Kita, S.; Nakatsu, Y.; Aoki, N.; Mori, Y.; Maenaka, K.; Takeda, M.; Kidokoro, M. Heat shock protein 70 regulates degradation of the mumps virus phosphoprotein via the ubiquitin-proteasome pathway. J. Virol. 2015, 89, 3188–3199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, C.; Cao, Y.; Jamal, M.; Chen, X.; Zheng, J.; Li, L.; You, J.; Zhu, Q.; Liu, S.; et al. Rabies viruses leader RNA interacts with host Hsc70 and inhibits virus replication. Oncotarget 2017, 8, 43822–43837. [Google Scholar] [CrossRef] [Green Version]
- Lahaye, X.; Vidy, A.; Fouquet, B.; Blondel, D. Hsp70 Protein Positively Regulates Rabies Virus Infection. J. Virol. 2012, 86, 4743–4751. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.S.; Chase, M.A.; Senft, A.P.; Poynter, S.E.; Wong, H.R.; Page, K. Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 2009, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Baturcam, E.; Snape, N.; Yeo, T.H.; Schagen, J.; Thomas, E.; Logan, J.; Galbraith, S.; Collinson, N.; Phipps, S.; Fantino, E.; et al. Human Metapneumovirus Impairs Apoptosis of Nasal Epithelial Cells in Asthma via HSP70. J. Innate Immun. 2017, 9, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.P.; Simabuco, F.M.; Tamura, R.E.; Guerrero, M.C.; Ribeiro, P.G.G.; Libermann, T.A.; Zerbini, L.F.; Ventura, A.M. Human respiratory syncytial virus N, P and M protein interactions in HEK-293T cells. Virus Res. 2013, 177, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Oglesbee, M. Virus-heat shock protein interaction and a novel axis for innate antiviral immunity. Cells 2012, 1, 646–666. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P. Recruitment of Hsp70 chaperones: A crucial part of viral survival strategies. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 153, pp. 1–46. ISBN 0303-4240. [Google Scholar]
- Collins, P.L.; Hightower, L.E. Newcastle disease virus stimulates the cellular accumulation of stress (heat shock) mRNAs and proteins. J. Virol. 1982, 44, 703–707. [Google Scholar]
- Oglesbee, M.; Krakowka, S. Cellular stress response induces selective intranuclear trafficking and accumulation of morbillivirus major core protein. Lab. Investig. 1993, 68, 109–117. [Google Scholar] [PubMed]
- Brown, G.; Rixon, H.W.M.; Steel, J.; McDonald, T.P.; Pitt, A.R.; Graham, S.; Sugrue, R.J. Evidence for an association between heat shock protein 70 and the respiratory syncytial virus polymerase complex within lipid-raft membranes during virus infection. Virology 2005, 338, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, A.; Yeo, D.; Brown, G.; Myaing, M.Z.; Iyer, L.R.; Fleck, R.; Tan, B.-H.; Aitken, J.; Sanmun, D.; Tang, K.; et al. Protein analysis of purified respiratory syncytial virus particles reveals an important role for heat shock protein 90 in virus particle assembly. Mol. Cell. Proteom. 2010, 9, 1829–1848. [Google Scholar] [CrossRef]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional Characterization of Negri Bodies (NBs) in Rabies Virus-Infected Cells: Evidence that NBs Are Sites of Viral Transcription and Replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef] [Green Version]
- Oglesbee, M.J.; Kenney, H.; Kenney, T.; Krakowka, S. Enhanced production of morbillivirus gene-specific RNAs following induction of the cellular stress response in stable persistent infection. Virology 1993, 192, 556–567. [Google Scholar] [CrossRef]
- Oglesbee, M.J.; Liu, Z.; Kenney, H.; Brooks, C.L. The Highly Inducible Member of the 70 kDa Family of Heat Shock Proteins Increases Canine Distemper Virus Polymerase Activity. J. Gen. Virol. 1996, 77, 2125–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos, D.; Norrby, E.; Oglesbee, M. The cellular stress response increases measles virus-induced cytopathic effect. J. Gen. Virol. 1998, 79 Pt 7, 1769–1773. [Google Scholar] [CrossRef]
- Parks, C.L.; Lerch, R.A.; Walpita, P.; Sidhu, M.S.; Udem, S.A. Enhanced measles virus cDNA rescue and gene expression after heat shock. J. Virol. 1999, 73, 3560–3566. [Google Scholar] [PubMed]
- Heller, M.; Vasconcelos, D.; Cummins, J.; Oglesbee, M. Interferon-alpha inhibits the emergence of cellular stress response-dependent morbillivirus large plaque variants. Antivir. Res. 1998, 38, 195–207. [Google Scholar] [CrossRef]
- Vasconcelos, D.Y.; Cai, X.H.; Oglesbee, M.J. Constitutive overexpression of the major inducible 70 kDa heat shock protein mediates large plaque formation by measles virus. J. Gen. Virol. 1998, 79 Pt 9, 2239–2247. [Google Scholar] [CrossRef]
- Zhang, X.; Glendening, C.; Linke, H.; Parks, C.L.; Brooks, C.; Udem, S.A.; Oglesbee, M. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 2002, 76, 8737–8746. [Google Scholar] [CrossRef]
- Carsillo, T.; Traylor, Z.; Choi, C.; Niewiesk, S.; Oglesbee, M. hsp72, a host determinant of measles virus neurovirulence. J. Virol. 2006, 80, 11031–11039. [Google Scholar] [CrossRef]
- Kim, M.Y.; Ma, Y.; Zhang, Y.; Li, J.; Shu, Y.; Oglesbee, M. hsp70-Dependent Antiviral Immunity against Cytopathic Neuronal Infection by Vesicular Stomatitis Virus. J. Virol. 2013, 87, 10668–10678. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Bourhis, J.M.; Longhi, S.; Carsillo, T.; Buccellato, M.; Morin, B.; Canard, B.; Oglesbee, M. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 2005, 337, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Couturier, M.; Buccellato, M.; Costanzo, S.; Bourhis, J.-M.; Shu, Y.; Nicaise, M.; Desmadril, M.; Flaudrops, C.; Longhi, S.; Oglesbee, M. High affinity binding between Hsp70 and the C-terminal domain of the measles virus nucleoprotein requires an Hsp40 co-chaperone. J. Mol. Recognit. 2010, 23, 301–315. [Google Scholar] [CrossRef]
- Carsillo, T.; Zhang, X.; Vasconcelos, D.; Niewiesk, S.; Oglesbee, M. A single codon in the nucleocapsid protein C terminus contributes to in vitro and in vivo fitness of Edmonston measles virus. J. Virol. 2006, 80, 2904–2912. [Google Scholar] [CrossRef] [PubMed]
- García-Dorival, I.; Wu, W.; Armstrong, S.D.; Barr, J.N.; Carroll, M.W.; Hewson, R.; Hiscox, J.A. Elucidation of the Cellular Interactome of Ebola Virus Nucleoprotein and Identification of Therapeutic Targets. J. Proteome Res. 2016, 15, 4290–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, E.V.; Pacheco, J.R.; Hume, A.J.; Cressey, T.N.; Deflubé, L.R.; Ruedas, J.B.; Connor, J.H.; Ebihara, H.; Mühlberger, E. An RNA polymerase II-driven Ebola virus minigenome system as an advanced tool for antiviral drug screening. Antivir. Res. 2017, 146, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Tchesnokov, E.P.; Raeisimakiani, P.; Ngure, M.; Marchant, D.; Götte, M. Recombinant RNA-Dependent RNA Polymerase Complex of Ebola Virus. Sci. Rep. 2018, 8, 3970. [Google Scholar] [CrossRef] [PubMed]
- Munday, D.C.; Wu, W.; Smith, N.; Fix, J.; Noton, S.L.; Galloux, M.; Touzelet, O.; Armstrong, S.D.; Dawson, J.M.; Aljabr, W.; et al. Interactome Analysis of the Human Respiratory Syncytial Virus RNA Polymerase Complex Identifies Protein Chaperones as Important Cofactors That Promote L-Protein Stability and RNA Synthesis. J. Virol. 2015, 89, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Kubota, T.; Nakatsu, Y.; Tahara, M.; Kidokoro, M.; Takeda, M. Heat Shock Protein 90 Ensures Efficient Mumps Virus Replication by Assisting with Viral Polymerase Complex Formation. J. Virol. 2017, 91, e02220-16. [Google Scholar] [CrossRef]
- Sztuba-Solinska, J.; Diaz, L.; Kumar, M.R.; Kolb, G.; Wiley, M.R.; Jozwick, L.; Kuhn, J.H.; Palacios, G.; Radoshitzky, S.R.; Le Grice, S.F.J.; et al. A small stem-loop structure of the Ebola virus trailer is essential for replication and interacts with heat-shock protein A8. Nucleic Acids Res. 2016, 44, 9831–9846. [Google Scholar] [CrossRef]
- Wu, W.; Tran, K.C.; Teng, M.N.; Heesom, K.J.; Matthews, D.A.; Barr, J.N.; Hiscox, J.A. The interactome of the human respiratory syncytial virus NS1 protein highlights multiple effects on host cell biology. J. Virol. 2012, 86, 7777–7789. [Google Scholar] [CrossRef]
- Spurgers, K.B.; Alefantis, T.; Peyser, B.D.; Ruthel, G.T.; Bergeron, A.A.; Costantino, J.A.; Enterlein, S.; Kota, K.P.; Boltz, R.C.D.; Aman, M.J.; et al. Identification of essential filovirion-associated host factors by serial proteomic analysis and RNAi screen. Mol. Cell. Proteom. 2010, 9, 2690–2703. [Google Scholar] [CrossRef]
- Liang, J.; Sagum, C.A.; Bedford, M.T.; Sidhu, S.S.; Sudol, M.; Han, Z.; Harty, R.N. Chaperone-Mediated Autophagy Protein BAG3 Negatively Regulates Ebola and Marburg VP40-Mediated Egress. PLoS Pathog. 2017, 13, e1006132. [Google Scholar] [CrossRef]
- Liang, B.; Li, Z.; Jenni, S.; Rahmeh, A.A.; Morin, B.M.; Grant, T.; Grigorieff, N.; Harrison, S.C.; Whelan, S.P.J. Structure of the L Protein of Vesicular Stomatitis Virus from Electron Cryomicroscopy. Cell 2015, 162, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Bloyet, L.-M.; Welsch, J.; Enchery, F.; Mathieu, C.; de Breyne, S.; Horvat, B.; Grigorov, B.; Gerlier, D. HSP90 Chaperoning in Addition to Phosphoprotein Required for Folding but Not for Supporting Enzymatic Activities of Measles and Nipah Virus L Polymerases. J. Virol. 2016, 90, 6642–6656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, R.; Andino, R.; Frydman, J. Hsp90 inhibitors exhibit resistance-free antiviral activity against respiratory syncytial virus. PLoS ONE 2013, 8, e56762. [Google Scholar] [CrossRef]
- Connor, J.H.; McKenzie, M.O.; Parks, G.D.; Lyles, D.S. Antiviral activity and RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology 2007, 362, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, F.; Liu, J.; Wang, D.; Yan, Y.; Ji, S.; Zan, J.; Zhou, J. The co-chaperone Cdc37 regulates the rabies virus phosphoprotein stability by targeting to Hsp90AA1 machinery. Sci. Rep. 2016, 6, 27123. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, X.; Zan, J.; Wu, Y.; Ye, C.; Ruan, X.; Zhou, J. Cellular Chaperonin CCT Contributes to Rabies Virus Replication during Infection. J. Virol. 2013, 87, 7608–7621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ye, C.; Ruan, X.; Zan, J.; Xu, Y.; Liao, M.; Zhou, J. The chaperonin CCTα is required for efficient transcription and replication of rabies virus. Microbiol. Immunol. 2014, 58, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roobol, A.; Sahyoun, Z.P.; Carden, M.J. Selected subunits of the cytosolic chaperonin associate with microtubules assembled in vitro. J. Biol. Chem. 1999, 274, 2408–2415. [Google Scholar] [CrossRef]
- García-Dorival, I.; Wu, W.; Dowall, S.; Armstrong, S.; Touzelet, O.; Wastling, J.; Barr, J.N.; Matthews, D.; Carroll, M.; Hewson, R.; et al. Elucidation of the Ebola virus VP24 cellular interactome and disruption of virus biology through targeted inhibition of host-cell protein function. J. Proteome Res. 2014, 13, 5120–5135. [Google Scholar] [CrossRef]
- Roux, L. Selective and transient association of Sendai virus HN glycoprotein with BiP. Virology 1990, 175, 161–166. [Google Scholar] [CrossRef]
- Watowich, S.S.; Morimoto, R.I.; Lamb, R. A Flux of the paramyxovirus hemagglutinin-neuraminidase glycoprotein through the endoplasmic reticulum activates transcription of the GRP78-BiP gene. J. Virol. 1991, 65, 3590–3597. [Google Scholar] [PubMed]
- Bitko, V.; Barik, S. An endoplasmic reticulum-specific stress-activated caspase (caspase-12) is implicated in the apoptosis of A549 epithelial cells by respiratory syncytial virus. J. Cell. Biochem. 2001, 80, 441–454. [Google Scholar] [CrossRef]
- Bolt, G. The measles virus (MV) glycoproteins interact with cellular chaperones in the endoplasmic reticulum and MV infection upregulates chaperone expression. Arch. Virol. 2001, 146, 2055–2068. [Google Scholar] [CrossRef] [PubMed]
- Patrick Reid, S.; Shurtleff, A.C.; Costantino, J.A.; Tritsch, S.R.; Retterer, C.; Spurgers, K.B.; Bavari, S. HSPA5 is an essential host factor for Ebola virus infection. Antivir. Res. 2014, 109, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.T.; Randall, R.E.; Lamb, R.A. Intracellular maturation and transport of the SV5 type II glycoprotein hemagglutinin-neuraminidase: Specific and transient association with GRP78-BiP in the endoplasmic reticulum and extensive internalization from the cell surface. J. Cell Biol. 1989, 109, 3273–3289. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.T.; Hiebert, S.W.; Lamb, R.A. Different roles of individual N-linked oligosaccharide chains in folding, assembly, and transport of the simian virus 5 hemagglutinin-neuraminidase. Mol. Cell. Biol. 1990, 10, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Stott, E.J.; Wertz, G.W. Intracellular processing of the human respiratory syncytial virus fusion glycoprotein: Amino acid substitutions affecting folding, transport and cleavage. J. Gen. Virol. 1992, 73 Pt 5, 1177–1188. [Google Scholar] [CrossRef]
- Gaudin, Y. Folding of rabies virus glycoprotein: Epitope acquisition and interaction with endoplasmic reticulum chaperones. J. Virol. 1997, 71, 3742–3750. [Google Scholar]
- Booth, L.; Roberts, J.L.; Ecroyd, H.; Tritsch, S.R.; Bavari, S.; Reid, S.P.; Proniuk, S.; Zukiwski, A.; Jacob, A.; Sepúlveda, C.S.; et al. AR-12 Inhibits Multiple Chaperones Concomitant With Stimulating Autophagosome Formation Collectively Preventing Virus Replication. J. Cell. Physiol. 2016, 231, 2286–2302. [Google Scholar] [CrossRef]
- Roberts, J.L.; Tavallai, M.; Nourbakhsh, A.; Fidanza, A.; Cruz-Luna, T.; Smith, E.; Siembida, P.; Plamondon, P.; Cycon, K.A.; Doern, C.D.; et al. GRP78/Dna K Is a Target for Nexavar/Stivarga/Votrient in the Treatment of Human Malignancies, Viral Infections and Bacterial Diseases. J. Cell. Physiol. 2015, 230, 2552–2578. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Cash, D.R.; Tavallai, S.; Jean, S.; Fidanza, A.; Cruz-Luna, T.; Siembiba, P.; Cycon, K.A.; Cornelissen, C.N.; et al. GRP78/BiP/HSPA5/Dna K is a universal therapeutic target for human disease. J. Cell. Physiol. 2015, 230, 1661–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wang, Y.; Frabutt, D.A.; Zhang, X.; Yao, X.; Hu, D.; Zhang, Z.; Liu, C.; Zheng, S.; Xiang, S.-H.; et al. Mechanistic understanding of N-glycosylation in Ebola virus glycoprotein maturation and function. J. Biol. Chem. 2017, 292, 5860–5870. [Google Scholar] [CrossRef]
- Hammond, C.; Helenius, A. Folding of VSV G protein: Sequential interaction with BiP and calnexin. Science 1994, 266, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Ora, A.; Van, P.N.; Helenius, A. Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Mol. Biol. Cell 1995, 6, 1173–1184. [Google Scholar] [CrossRef]
- Bloor, S.; Maelfait, J.; Krumbach, R.; Beyaert, R.; Randow, F. Endoplasmic reticulum chaperone gp96 is essential for infection with vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2010, 107, 6970–6975. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.R.; McCarthy, S.; Chrovian, A.; Olinger, G.; Stossel, A.; Geisbert, T.W.; Hensley, L.E.; Connor, J.H. Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antivir. Res. 2010, 87, 187–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Erlichman, C.; McDonald, C.J.; Ingle, J.N.; Zollman, P.; Iankov, I.; Russell, S.J.; Galanis, E. Heat shock protein inhibitors increase the efficacy of measles virotherapy. Gene Ther. 2008, 15, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Munday, D.C.; Howell, G.; Barr, J.N.; Hiscox, J.A. Proteomic analysis of mitochondria in respiratory epithelial cells infected with human respiratory syncytial virus and functional implications for virus and cell biology. J. Pharm. Pharmacol. 2015, 67, 300–318. [Google Scholar] [CrossRef]
- Geller, R.; Vignuzzi, M.; Andino, R.; Frydman, J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007, 21, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Okudera, M.; Gojoubori, T.; Tsujino, I.; Asano, M. Effect of ionomycin on interaction of calnexin with vesicular stomatitis virus glycoprotein is cell type-specific. J. Oral Sci. 2015, 57, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Warren, T.K.; Zhao, X.; Gill, T.; Guo, F.; Wang, L.; Comunale, M.A.; Du, Y.; Alonzi, D.S.; Yu, W.; et al. Small molecule inhibitors of ER α-glucosidases are active against multiple hemorrhagic fever viruses. Antivir. Res. 2013, 98, 432–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, M.; Vandenbroeck, K. The endoplasmic reticulum protein folding factory and its chaperones: New targets for drug discovery? Br. J. Pharmacol. 2011, 162, 328–345. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Shu, Y.; Carsillo, T.; Zhang, J.; Yu, L.; Peterson, C.; Longhi, S.; Girod, S.; Niewiesk, S.; Oglesbee, M. hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J. Virol. 2013, 87, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Carsillo, T.; Carsillo, M.; Niewiesk, S.; Vasconcelos, D.; Oglesbee, M. Hyperthermic pre-conditioning promotes measles virus clearance from brain in a mouse model of persistent infection. Brain Res. 2004, 1004, 73–82. [Google Scholar] [CrossRef]
- Pockley, A.G.; Henderson, B. Extracellular cell stress (heat shock) proteins-immune responses and disease: An overview. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Munday, D.C.; Hiscox, J.A.; Barr, J.N. Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus subgroup B using SILAC coupled to LC-MS/MS. Proteom. 2010, 10, 4320–4334. [Google Scholar] [CrossRef] [PubMed]
- Taguwa, S.; Maringer, K.; Li, X.; Bernal-Rubio, D.; Rauch, J.N.; Gestwicki, J.E.; Andino, R.; Fernandez-Sesma, A.; Frydman, J. Defining Hsp70 Subnetworks in Dengue Virus Replication Reveals Key Vulnerability in Flavivirus Infection. Cell 2015, 163, 1108–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Wang, T.; Araujo, T.L.S.; Sharma, S.; Brodsky, J.L.; Chiosis, G. Adapting to stress—Chaperome networks in cancer. Nat. Rev. Cancer 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Family | Virus | Chaperone Target | Compound 1 | Reference(s) |
|---|---|---|---|---|
| Filoviridae | EBOV | Hsp90 | 17AAG, GA, Radicicol, SNX 9503/2113/7023/7021 | [110] |
| Hsp70 | RNA interference | [103] | ||
| VER155008 | [76] | |||
| JG40 | [77] | |||
| DNAJB2 | RNA interference | [83] | ||
| Multiple chaperones | EGCG | [98] | ||
| BiP | RNA interference | [83,98,103] | ||
| Hsc70 | RNA interference | [81] | ||
| MARV | BiP | RNA interference | [83] | |
| Paramyxoviridae | MuV | Hsp90 | 17AAG | [80] |
| Hsp70 | VER155008 + 17AAG | [80] | ||
| BiP + Hsp27 | RNA interference | [103] | ||
| Multiple chaperones | Sorafenib, Sorafenib + Sildenafil, Sildenafil + AR-12 | [104,105] | ||
| MeV | Hsp90 | GA, 17DMAG | [86,111] | |
| RNA interference | [86] | |||
| Hsp70 | VER155008 + 17AAG | [80] | ||
| BiP + Hsp27 | RNA interference | [103] | ||
| Multiple chaperones | Sildenafil + AR-12 | [105] | ||
| PIV2 | Hsp90 | GA | [88] | |
| SV5 | Hsp90 | GA/Radicicol | [88] | |
| Pneumoviridae | RSV | Hsp90 | GA, 17AAG, 17DMAG | [62,79,87] |
| RNA interference | [62] | |||
| Hsp70 | VER155008, PIF, MKT007, YM1 | [79] | ||
| Hsc70 | RNA interference | [62] | ||
| MPV | Hsp70 | VER155008 | [55] | |
| Rhabdoviridae | RABV | Hsp90 | 17AAG | [89] |
| RNA interference | [89] | |||
| Hsp90/Cdc37 | Celastrol | [89] | ||
| Cdc37 | RNA interference | [89] | ||
| Hsp70 | RNA interference | [53] | ||
| CCTγ | RNA interference | [90] | ||
| CCTα | RNA interference | [91] | ||
| Block Hsp induction | Quercetin | [53] | ||
| Multiple chaperones | Sorafenib, Sorafenib + Sildenafil | [105] | ||
| VSV | Hsp90 | GA | [88] | |
| RNA interference | [88] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latorre, V.; Mattenberger, F.; Geller, R. Chaperoning the Mononegavirales: Current Knowledge and Future Directions. Viruses 2018, 10, 699. https://doi.org/10.3390/v10120699
Latorre V, Mattenberger F, Geller R. Chaperoning the Mononegavirales: Current Knowledge and Future Directions. Viruses. 2018; 10(12):699. https://doi.org/10.3390/v10120699
Chicago/Turabian StyleLatorre, Victor, Florian Mattenberger, and Ron Geller. 2018. "Chaperoning the Mononegavirales: Current Knowledge and Future Directions" Viruses 10, no. 12: 699. https://doi.org/10.3390/v10120699
APA StyleLatorre, V., Mattenberger, F., & Geller, R. (2018). Chaperoning the Mononegavirales: Current Knowledge and Future Directions. Viruses, 10(12), 699. https://doi.org/10.3390/v10120699