Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus and Plasmids
2.2. Transfection and Reagents
2.3. Co-Immunoprecipitation (Co-IP) and GST-Pulldown Assay
2.4. Immunoblotting
2.5. Luciferase Assay
2.6. Reverse-Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.7. Confocal Microscopy
2.8. Statistical Analysis
3. Results
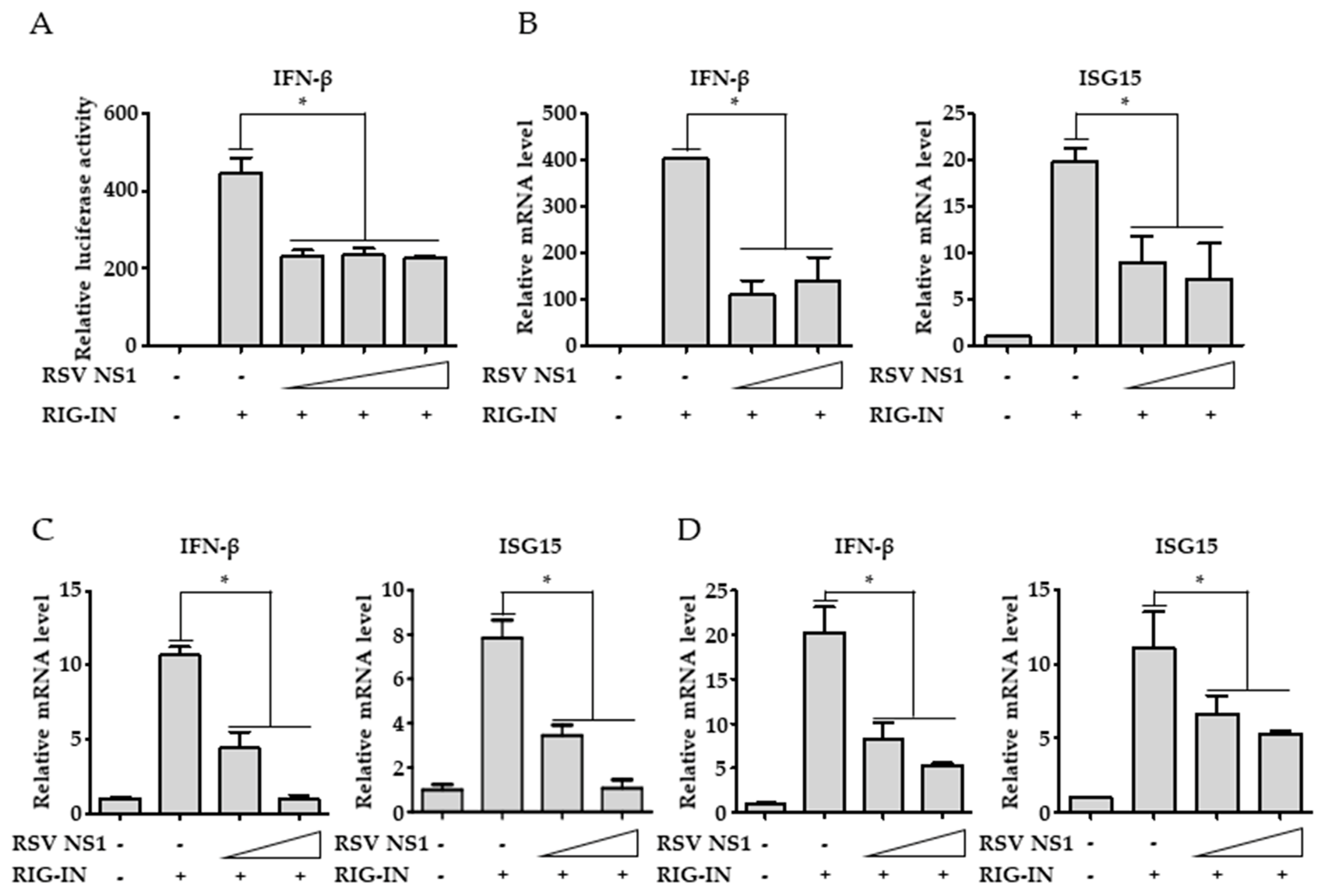
3.1. RSV NS1 Inhibits RIG-I-Mediated Interferon Signaling
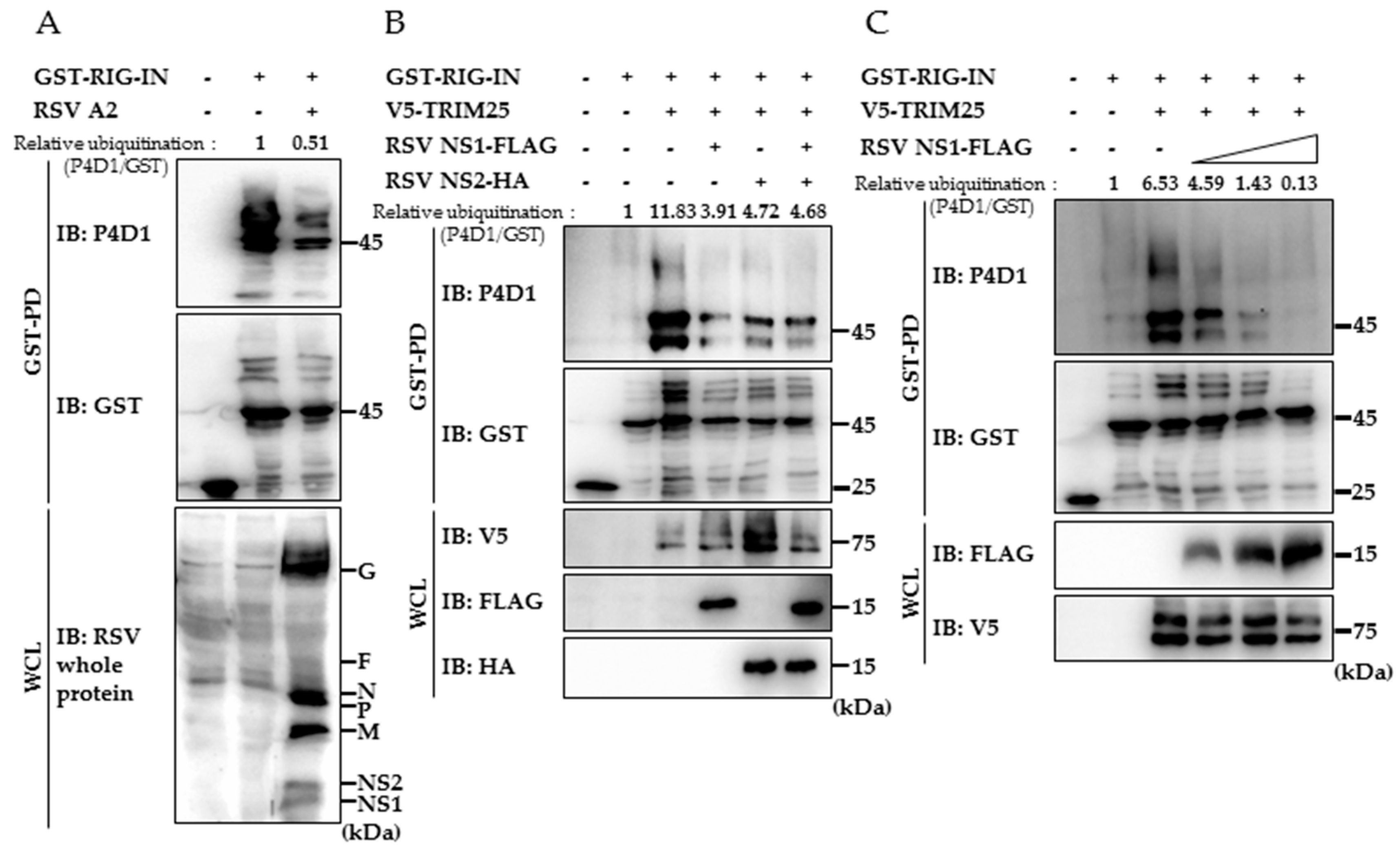
3.2. RSV NS1 Suppresses TRIM25-Mediated RIG-I Ubiquitination
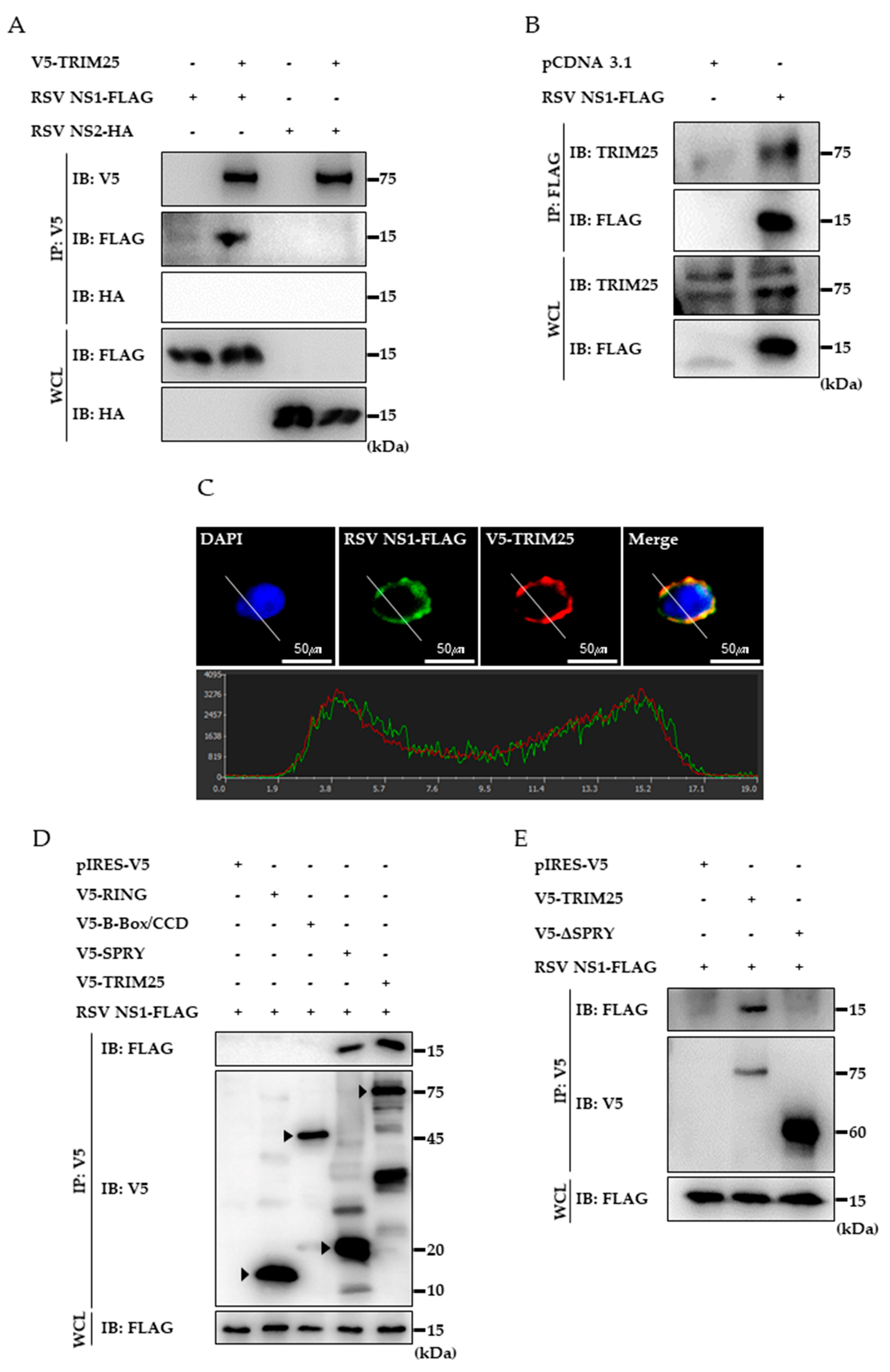
3.3. RSV NS1 Protein Interacts with TRIM25
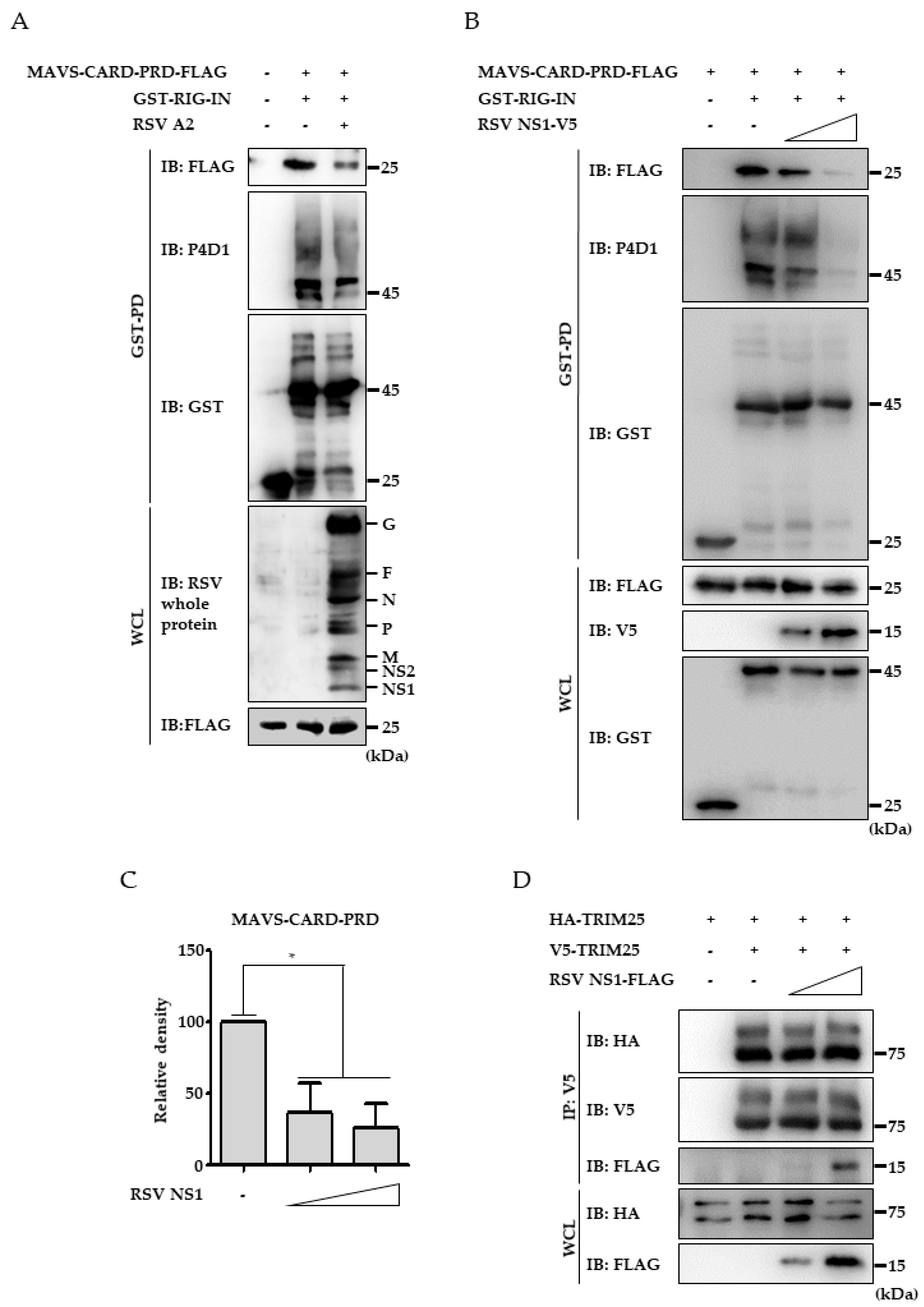
3.4. RSV NS1 Protein Hinders RIG-I 2CARD Interaction with MAVS CARD
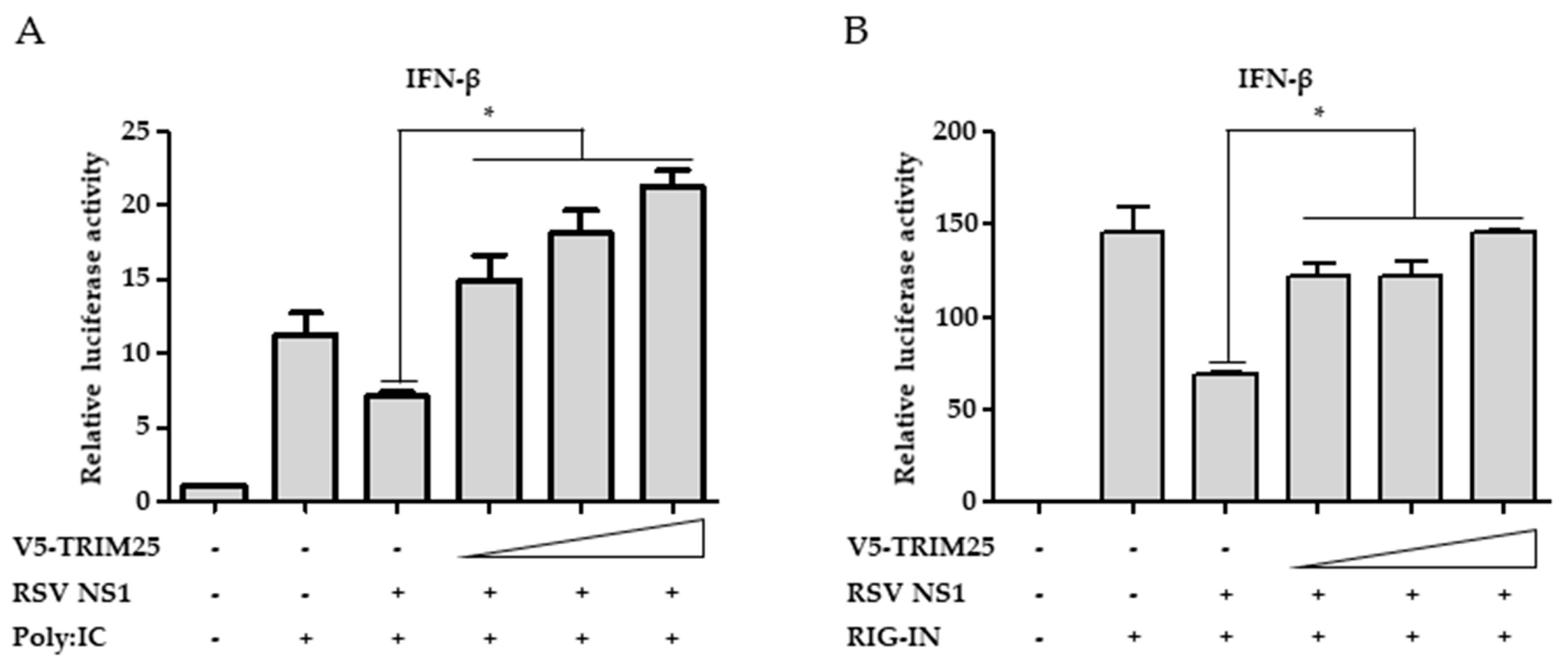
3.5. Ectopic Expression of TRIM25 Reverses the Effect of NS1 on RIG-I-Mediated Interferon Signaling
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Tschopp, J.; Karin, M. Intracellular pattern recognition receptors in the host response. Nature 2006, 442, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, V.G.; Sun, Q.; Bhoj, E.J.; Somers, C.; Chen, X.; Torres, J.P.; Mejias, A.; Gomez, A.M.; Jafri, H.; Ramilo, O.; et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2008, 105, 14046–14051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Martin-Vicente, M.; Medrano, L.M.; Resino, S.; Garcia-Sastre, A.; Martinez, I. TRIM25 in the Regulation of the Antiviral Innate Immunity. Front. Immunol. 2017, 8, 1187. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Canedo-Marroquin, G.; Acevedo-Acevedo, O.; Rey-Jurado, E.; Saavedra, J.M.; Lay, M.K.; Bueno, S.M.; Riedel, C.A.; Kalergis, A.M. Modulation of Host Immunity by Human Respiratory Syncytial Virus Virulence Factors: A Synergic Inhibition of Both Innate and Adaptive Immunity. Front. Cell. Infect. Microbiol. 2017, 7, 367. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, J.; Zheng, K.; Hou, Y.; Zhao, F.; Zhao, D. Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology 2014, 57, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, L.; Wang, H.; Zhang, G.; Sun, X. Respiratory syncytial virus non-structural protein 1 facilitates virus replication through miR-29a-mediated inhibition of interferon-alpha receptor. Biochem. Biophys. Res. Commun. 2016, 478, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yang, P.; Tang, Y.; Pan, Z.; Zhao, D. Respiratory Syncytial Virus Nonstructural Proteins Upregulate SOCS1 and SOCS3 in the Different Manner from Endogenous IFN Signaling. J. Immunol. Res. 2015, 2015, 738547. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef] [PubMed]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 2012, 7, e29386. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Majumdar, T.; Dhar, J.; Chattopadhyay, S.; Bandyopadhyay, S.K.; Verbovetskaya, V.; Sen, G.C.; Barik, S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013, 23, 1025–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swedan, S.; Andrews, J.; Majumdar, T.; Musiyenko, A.; Barik, S. Multiple functional domains and complexes of the two nonstructural proteins of human respiratory syncytial virus contribute to interferon suppression and cellular location. J. Virol. 2011, 85, 10090–10100. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Heo, J.; Yi, C.M.; Ban, J.; Lee, N.J.; Lee, N.R.; Kim, S.W.; Kim, N.J.; Inn, K.S. A novel p38 mitogen activated protein kinase (MAPK) specific inhibitor suppresses respiratory syncytial virus and influenza A virus replication by inhibiting virus-induced p38 MAPK activation. Biochem. Biophys. Res. Commun. 2016, 477, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Kirchhofer, A.; Shin, Y.C.; Inn, K.S.; Liang, C.; Cui, S.; Myong, S.; Ha, T.; Hopfner, K.P.; Jung, J.U. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc. Natl. Acad. Sci. USA 2008, 105, 16743–16748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Inhibits Type I Interferon Production by Interfering with TRIM25-Mediated RIG-I Ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidere, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ban, J.; Lee, N.-R.; Lee, N.-J.; Lee, J.K.; Quan, F.-S.; Inn, K.-S. Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling. Viruses 2018, 10, 716. https://doi.org/10.3390/v10120716
Ban J, Lee N-R, Lee N-J, Lee JK, Quan F-S, Inn K-S. Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling. Viruses. 2018; 10(12):716. https://doi.org/10.3390/v10120716
Chicago/Turabian StyleBan, Junsu, Na-Rae Lee, Noh-Jin Lee, Jong Kil Lee, Fu-Shi Quan, and Kyung-Soo Inn. 2018. "Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling" Viruses 10, no. 12: 716. https://doi.org/10.3390/v10120716
APA StyleBan, J., Lee, N. -R., Lee, N. -J., Lee, J. K., Quan, F. -S., & Inn, K. -S. (2018). Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling. Viruses, 10(12), 716. https://doi.org/10.3390/v10120716