All-Round Manipulation of the Actin Cytoskeleton by HIV



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inbound vs. Outbound HIV
3. Cell-Cell Transfer of HIV
3.1. The Virological Synapse
3.2. The Infectious Synapse
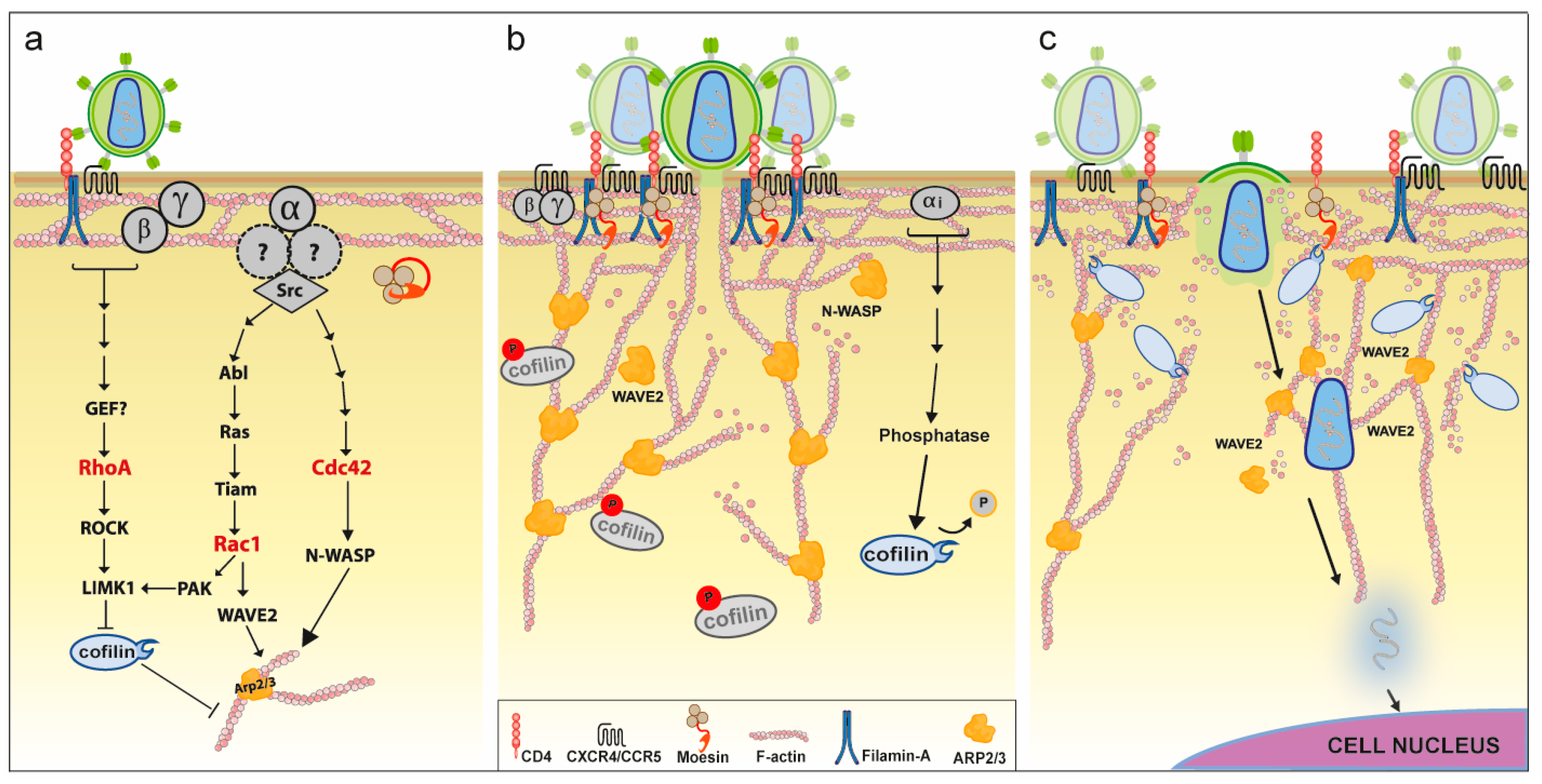
4. Direct Association between HIV and F-Actin
4.1. Actin in HIV Virions
4.2. Physical Interaction between Gag and F-Actin
4.3. Role of Actin for HIV Entry
4.4. Role of Actin for Free-Virus Egress
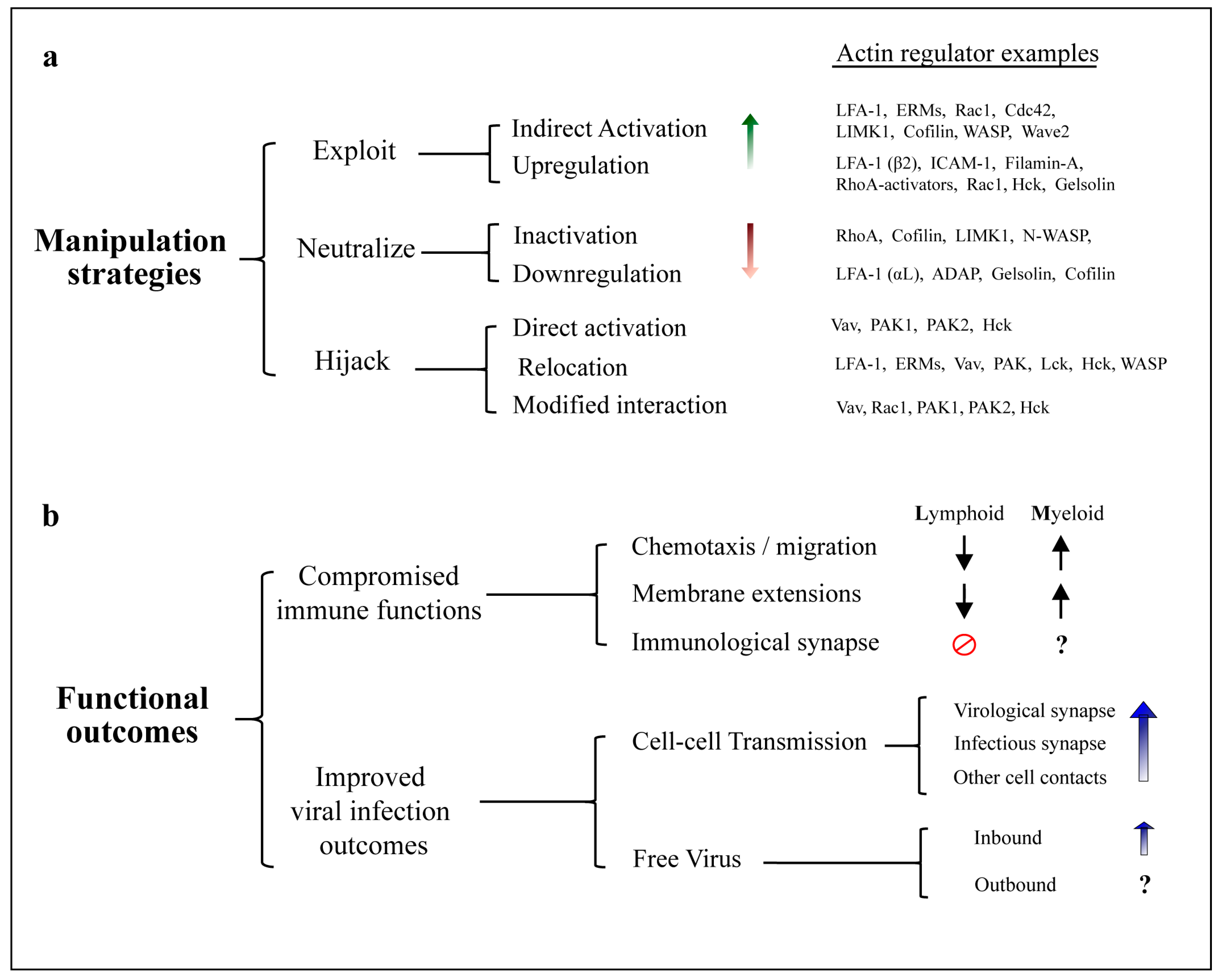
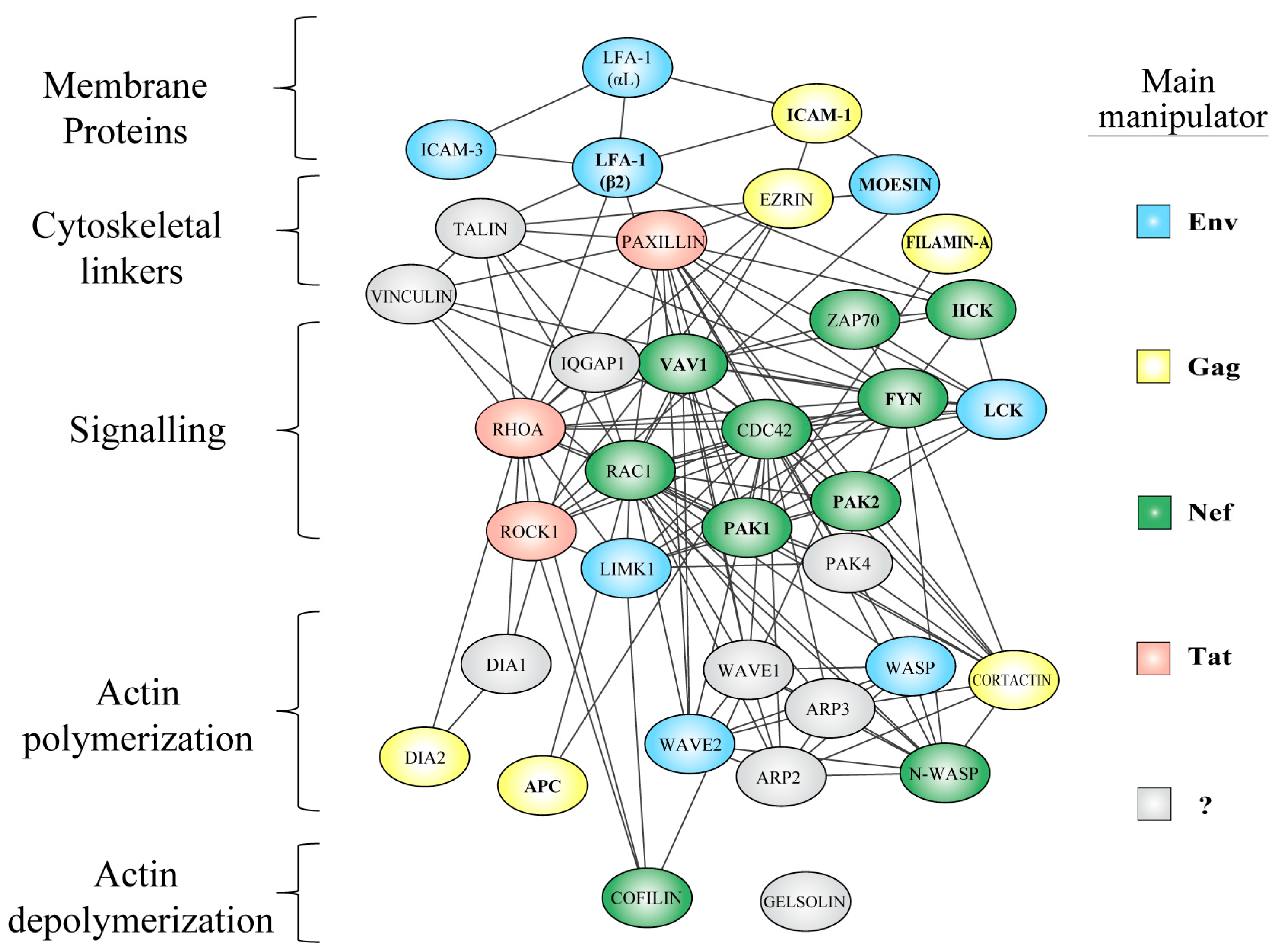
5. Manipulation of the Actin Cytoskeleton
5.1. Exploitation of Actin Regulators and Pathways by HIV
5.1.1. Membrane-Cytoskeleton Linkers
5.1.2. Rho-GTPase Signaling
5.1.3. Actin Polymerizing Factors
5.1.4. Actin Depolymerizing Factors
5.2. Neutralization of Actin Regulators by HIV
5.2.1. RhoA
5.2.2. Cofilin
5.2.3. N-WASP
5.3. Actin Regulators Hijacked by HIV
5.3.1. Vav
5.3.2. PAK
5.3.3. Hck
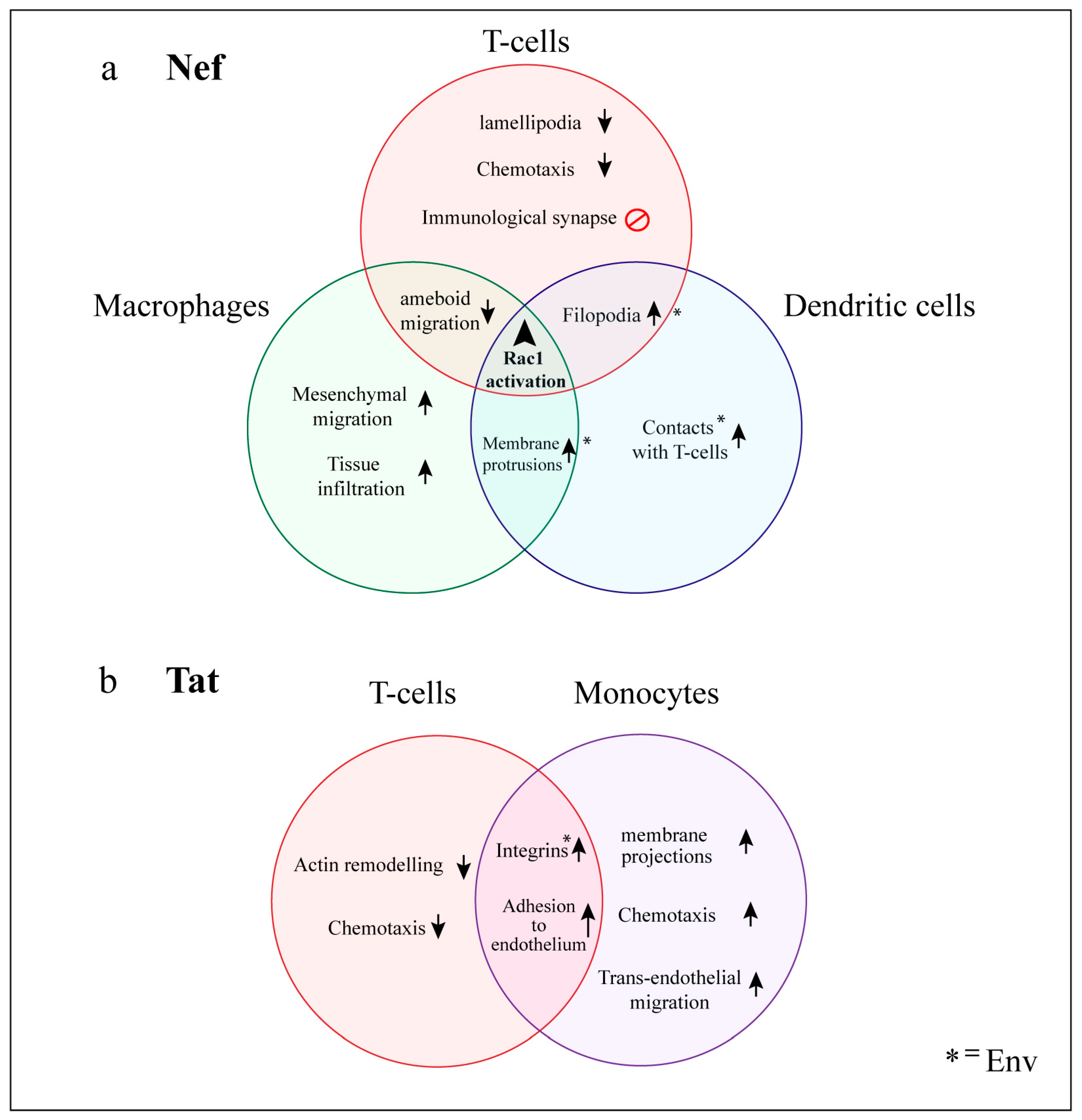
6. Functional Consequences of HIV Manipulation of Actin Networks
6.1. T-cells
6.1.1. Impairment of the Immunological Synapse
6.1.2. Virological Synapse Formation
6.2. Myeloid Cells
7. Conclusions and Perspective
Acknowledgments
Conflicts of Interest
References
- Povarova, O.I.; Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K. Actinous enigma or enigmatic actin: Folding, structure, and functions of the most abundant eukaryotic protein. Intrinsically Disord. Proteins 2014, 2, e34500. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 2011, 9, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Wu, Y. Viral exploitation of actin: Force-generation and scaffolding functions in viral infection. Virol. Sin. 2014, 29, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Dramsi, S.; Cossart, P. Intracellular pathogens and the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 1998, 14, 137–166. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.D.; Way, M. Arp2/3-mediated actin-based motility: A tail of pathogen abuse. Cell Host Microbe 2013, 14, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Hildreth, J.E.; Schwartz, D.H. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J. Virol. 1998, 72, 5251–5255. [Google Scholar] [PubMed]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, W.; Yin, W.; Guo, J.; Zhang, Z.P.; Zeng, D.; Zhang, X.; Wu, Y.; Zhang, X.E.; Cui, Z. Single-Particle Tracking of Human Immunodeficiency Virus Type 1 Productive Entry into Human Primary Macrophages. ACS Nano 2017, 11, 3890–3903. [Google Scholar] [CrossRef] [PubMed]
- Bukrinskaya, A.; Brichacek, B.; Mann, A.; Stevenson, M. Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J. Exp. Med. 1998, 188, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N.; Genovesio, A.; Kim, K.A.; Miko, S.; Perret, E.; Olivo-Marin, J.C.; Shorte, S.; Charneau, P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 2006, 3, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Guo, J.; Turner, A.; Yu, D.; Wang, W.; Meltzer, B.; He, S.; Hu, X.; Shang, H.; Kuhn, J.; et al. HIV-1 triggers WAVE2 phosphorylation in primary CD4 T cells and macrophages, mediating Arp2/3-dependent nuclear migration. J. Biol. Chem. 2014, 289, 6949–6959. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.U.; Saleh, S.; Sallmann, G.; Solomon, A.; Wightman, F.; Evans, V.A.; Boucher, G.; Haddad, E.K.; Sekaly, R.P.; Harman, A.N.; et al. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2010, 107, 16934–16939. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, K.; Nishi, M.; Matsunaga, S.; Okayama, A.; Anraku, M.; Kudoh, A.; Hirano, H.; Kimura, H.; Morikawa, Y.; Yamamoto, N.; et al. The tumour suppressor APC promotes HIV-1 assembly via interaction with Gag precursor protein. Nat. Commun. 2017, 8, 14259. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Ozaki, H.; Karaki, H.; Nonomura, Y. Actin filaments play an essential role for transport of nascent HIV-1 proteins in host cells. Biochem. Biophys. Res. Commun. 2004, 316, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Liu, L.; Woodruff, E.A.; Taylor, H.E.; Goodwin, J.S.; D’Aquila, R.T.; Spearman, P.; Hildreth, J.E.; Dong, X. Filamin A protein interacts with human immunodeficiency virus type 1 Gag protein and contributes to productive particle assembly. J. Biol. Chem. 2011, 286, 28498–28510. [Google Scholar] [CrossRef] [PubMed]
- Kerviel, A.; Thomas, A.; Chaloin, L.; Favard, C.; Muriaux, D. Virus assembly and plasma membrane domains: Which came first? Virus Res. 2013, 171, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Mitar, I.; Sattentau, Q.J. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 2007, 81, 5547–5560. [Google Scholar] [CrossRef] [PubMed]
- Gladnikoff, M.; Shimoni, E.; Gov, N.S.; Rousso, I. Retroviral assembly and budding occur through an actin-driven mechanism. Biophys. J. 2009, 97, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Ding, L.; Wang, J.J.; Qi, M.; Hammonds, J.; Chu, H.; Chen, X.; Hunter, E.; Spearman, P. ROCK1 and LIM kinase modulate retrovirus particle release and cell-cell transmission events. J. Virol. 2014, 88, 6906–6921. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nakamura, M.; Ohno, T.; Matsuda, Y.; Yuda, Y.; Nonomura, Y. Myosin-actin interaction plays an important role in human immunodeficiency virus type 1 release from host cells. Proc. Natl. Acad. Sci. USA 1995, 92, 2026–2030. [Google Scholar] [CrossRef] [PubMed]
- Audoly, G.; Popoff, M.R.; Gluschankof, P. Involvement of a small GTP binding protein in HIV-1 release. Retrovirology 2005, 2, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, M.; Nikolic, D.S.; Piguet, V. How HIV-1 takes advantage of the cytoskeleton during replication and cell-to-cell transmission. Viruses 2011, 3, 1757–1776. [Google Scholar] [CrossRef] [PubMed]
- Sodeik, B. Unchain my heart, baby let me go—The entry and intracellular transport of HIV. J. Cell Biol. 2002, 159, 393–395. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Fackler, O.T. How HIV takes advantage of the cytoskeleton in entry and replication. Viruses 2011, 3, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Debaisieux, S.; Rayne, F.; Yezid, H.; Beaumelle, B. The ins and outs of HIV-1 Tat. Traffic 2012, 13, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Olivetta, E.; Arenaccio, C.; Manfredi, F.; Anticoli, S.; Federico, M. The Contribution of Extracellular Nef to HIV-Induced Pathogenesis. Curr. Drug Targets 2016, 17, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.K.; Cruikshank, W.W.; Raina, J.; Blanchard, G.C.; Adler, W.H.; Walker, J.; Kornfeld, H. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J. Acquir. Immune Defic. Syndr. 1992, 5, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.U.; Freudenthal, P.S.; Barker, J.M.; Gezelter, S.; Inaba, K.; Steinman, R.M. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science 1992, 257, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.M.; Hocking, H.; Li, P.; Burrell, C.J. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 1999, 265, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Pearce-Pratt, R.; Phillips, D.M. Studies of adhesion of lymphocytic cells: Implications for sexual transmission of human immunodeficiency virus. Biol. Reprod. 1993, 48, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Bourinbaiar, A.S.; Phillips, D.M. Transmission of human immunodeficiency virus from monocytes to epithelia. J. Acquir. Immune Defic. Syndr. 1991, 4, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Perelson, A.S.; Kirschner, D.E.; de Boer, R. Dynamics of HIV infection of CD4+ T cells. Math. Biosci. 1993, 114, 81–125. [Google Scholar] [CrossRef]
- Zhang, C.; Zhou, S.; Groppelli, E.; Pellegrino, P.; Williams, I.; Borrow, P.; Chain, B.M.; Jolly, C. Hybrid spreading mechanisms and T cell activation shape the dynamics of HIV-1 infection. PLoS Comput. Biol. 2015, 11, e1004179. [Google Scholar] [CrossRef] [PubMed]
- Ganor, Y.; Zhou, Z.; Tudor, D.; Schmitt, A.; Vacher-Lavenu, M.C.; Gibault, L.; Thiounn, N.; Tomasini, J.; Wolf, J.P.; Bomsel, M. Within 1 h, HIV-1 uses viral synapses to enter efficiently the inner, but not outer, foreskin mucosa and engages Langerhans-T cell conjugates. Mucosal Immunol. 2010, 3, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Barreto-de-Souza, V.; Arakelyan, A.; Zicari, S.; Margolis, L.; Vanpouille, C. Monocytes but Not Lymphocytes Carrying HIV-1 on Their Surface Transmit Infection to Human Tissue Ex Vivo. J. Virol. 2016, 90, 9833–9840. [Google Scholar] [CrossRef] [PubMed]
- Sodora, D.L.; Gettie, A.; Miller, C.J.; Marx, P.A. Vaginal transmission of SIV: Assessing infectivity and hormonal influences in macaques inoculated with cell-free and cell-associated viral stocks. AIDS Res. Hum. Retrovir. 1998, 14 (Suppl. S1), S119–S123. [Google Scholar] [PubMed]
- Salle, B.; Brochard, P.; Bourry, O.; Mannioui, A.; Andrieu, T.; Prevot, S.; Dejucq-Rainsford, N.; Dereuddre-Bosquet, N.; Le Grand, R. Infection of macaques after vaginal exposure to cell-associated simian immunodeficiency virus. J. Infect. Dis. 2010, 202, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin-Gal, D.; Hulot, S.L.; Korioth-Schmitz, B.; Gombos, R.B.; Zheng, Y.; Owuor, J.; Lifton, M.A.; Ayeni, C.; Najarian, R.M.; Yeh, W.W.; et al. Efficiency of cell-free and cell-associated virus in mucosal transmission of human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 2013, 87, 13589–13597. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Murooka, T.T.; Imle, A.; Mempel, T.R. Adding new dimensions: Towards an integrative understanding of HIV-1 spread. Nat. Rev. Microbiol. 2014, 12, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Ladinsky, M.S.; Uchil, P.D.; Beloor, J.; Pi, R.; Herrmann, C.; Motamedi, N.; Murooka, T.T.; Brehm, M.A.; Greiner, D.L.; et al. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science 2015, 350, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Law, K.M.; Komarova, N.L.; Yewdall, A.W.; Lee, R.K.; Herrera, O.L.; Wodarz, D.; Chen, B.K. In Vivo HIV-1 Cell-to-Cell Transmission Promotes Multicopy Micro-compartmentalized Infection. Cell Rep. 2016, 15, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Casartelli, N. HIV-1 Cell-to-Cell Transmission and Antiviral Strategies: An Overview. Curr. Drug Targets 2016, 17, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Sattentau, Q.J. Retroviral spread by induction of virological synapses. Traffic 2004, 5, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus cell-to-cell transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef] [PubMed]
- Pearce-Pratt, R.; Malamud, D.; Phillips, D.M. Role of the cytoskeleton in cell-to-cell transmission of human immunodeficiency virus. J. Virol. 1994, 68, 2898–2905. [Google Scholar] [PubMed]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Plata, M.T.; Puigdomenech, I.; Izquierdo-Useros, N.; Puertas, M.C.; Carrillo, J.; Erkizia, I.; Clotet, B.; Blanco, J.; Martinez-Picado, J. The infectious synapse formed between mature dendritic cells and CD4(+) T cells is independent of the presence of the HIV-1 envelope glycoprotein. Retrovirology 2013, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.S.; Lehmann, M.; Felts, R.; Garcia, E.; Blanchet, F.P.; Subramaniam, S.; Piguet, V. HIV-1 activates Cdc42 and induces membrane extensions in immature dendritic cells to facilitate cell-to-cell virus propagation. Blood 2011, 118, 4841–4852. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C. T cell polarization at the virological synapse. Viruses 2010, 2, 1261–1278. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Sattentau, Q. Dangerous liaisons at the virological synapse. J. Clin. Investig. 2004, 114, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Fackler, O.T. HIV-1 at the immunological and T-lymphocytic virological synapse. Biol. Chem. 2008, 389, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Hubner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.; Li, X.D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 2009, 323, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, C.R. HIV-1 Trans Infection of CD4(+) T Cells by Professional Antigen Presenting Cells. Scientifica 2013, 2013, 164203. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.; Turville, S.G. Exploiting cellular contacts: Movement of HIV in the context of cellular communication. In Viral Transport, Assembly and Egress; Diefenbach, R., Cunningham, A.L., Eds.; Research Signpost: Kerala, India, 2011; Volume 1, pp. 133–151. [Google Scholar]
- Turville, S.G.; Santos, J.J.; Frank, I.; Cameron, P.U.; Wilkinson, J.; Miranda-Saksena, M.; Dable, J.; Stossel, H.; Romani, N.; Piatak, M., Jr.; et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood 2004, 103, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Turville, S.G.; Aravantinou, M.; Stossel, H.; Romani, N.; Robbiani, M. Resolution of de novo HIV production and trafficking in immature dendritic cells. Nat. Methods 2008, 5, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Iemma, T.L.; Shih, I.; Newsome, T.P.; McAllery, S.; Cunningham, A.L.; Turville, S.G. Mobilization of HIV spread by diaphanous 2 dependent filopodia in infected dendritic cells. PLoS Pathog. 2012, 8, e1002762. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.; Russell, R.A.; Sattentau, Q.J. High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. AIDS 2013, 27, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D. Dendritic Cells and HIV-1 Trans-Infection. Viruses 2010, 2, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Felts, R.L.; Narayan, K.; Estes, J.D.; Shi, D.; Trubey, C.M.; Fu, J.; Hartnell, L.M.; Ruthel, G.T.; Schneider, D.K.; Nagashima, K.; et al. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13336–13341. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Prasad, A.; Kuzontkoski, P.M.; Yu, J.; Groopman, J.E. Slit2N Inhibits Transmission of HIV-1 from Dendritic Cells to T-cells by Modulating Novel Cytoskeletal Elements. Sci. Rep. 2015, 5, 16833. [Google Scholar] [CrossRef] [PubMed]
- Peressin, M.; Proust, A.; Schmidt, S.; Su, B.; Lambotin, M.; Biedma, M.E.; Laumond, G.; Decoville, T.; Holl, V.; Moog, C. Efficient transfer of HIV-1 in trans and in cis from Langerhans dendritic cells and macrophages to autologous T lymphocytes. AIDS 2014, 28, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Neidleman, J.A.; Chen, J.C.; Kohgadai, N.; Muller, J.A.; Laustsen, A.; Thavachelvam, K.; Jang, K.S.; Sturzel, C.M.; Jones, J.J.; Ochsenbauer, C.; et al. Mucosal stromal fibroblasts markedly enhance HIV infection of CD4+ T cells. PLoS Pathog. 2017, 13, e1006163. [Google Scholar] [CrossRef] [PubMed]
- Sharova, N.; Swingler, C.; Sharkey, M.; Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. EMBO J. 2005, 24, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wolf, B.; Lamb, R.; Choppin, P.; Goldberg, A. The presence of actin in enveloped viruses. In Cell Motility; Goldman, R., Pollard, T., Rosenbaum, J., Eds.; Cold Spring Harbor Publications: Cold Spring Harbor, NY, USA, 1976; Volume 1, pp. 589–599. [Google Scholar]
- Ott, D.E.; Coren, L.V.; Kane, B.P.; Busch, L.K.; Johnson, D.G.; Sowder, R.C., 2nd; Chertova, E.N.; Arthur, L.O.; Henderson, L.E. Cytoskeletal proteins inside human immunodeficiency virus type 1 virions. J. Virol. 1996, 70, 7734–7743. [Google Scholar] [PubMed]
- Wilk, T.; Gowen, B.; Fuller, S.D. Actin associates with the nucleocapsid domain of the human immunodeficiency virus Gag polyprotein. J. Virol. 1999, 73, 1931–1940. [Google Scholar] [PubMed]
- Ott, D.E.; Coren, L.V.; Johnson, D.G.; Kane, B.P.; Sowder, R.C., 2nd; Kim, Y.D.; Fisher, R.J.; Zhou, X.Z.; Lu, K.P.; Henderson, L.E. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 2000, 266, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Katzav, S.; Martin-Zanca, D.; Barbacid, M. vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J. 1989, 8, 2283–2290. [Google Scholar] [PubMed]
- Linde, M.E.; Colquhoun, D.R.; Ubaida Mohien, C.; Kole, T.; Aquino, V.; Cotter, R.; Edwards, N.; Hildreth, J.E.; Graham, D.R. The conserved set of host proteins incorporated into HIV-1 virions suggests a common egress pathway in multiple cell types. J. Proteome Res. 2013, 12, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E. Cellular proteins detected in HIV-1. Rev. Med. Virol. 2008, 18, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E. Potential roles of cellular proteins in HIV-1. Rev. Med. Virol. 2002, 12, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, S.; Rahman, S.A.; de Marco, A.; Carlson, L.A.; Glass, B.; Oberwinkler, H.; Herold, N.; Briggs, J.A.; Muller, B.; Grunewald, K.; et al. The nucleocapsid domain of Gag is dispensable for actin incorporation into HIV-1 and for association of viral budding sites with cortical F-actin. J. Virol. 2014, 88, 7893–7903. [Google Scholar] [CrossRef] [PubMed]
- Turlure, F.; Devroe, E.; Silver, P.A.; Engelman, A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 3187–3208. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hashimoto, I.; Yamamoto, A.; Nishikawa, M.; Fujisawa, J.I. Rev-dependent association of the intron-containing HIV-1 gag mRNA with the nuclear actin bundles and the inhibition of its nucleocytoplasmic transport by latrunculin-B. Genes Cells 2000, 5, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, T.; Parra, M.; Vries, R.G.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar] [CrossRef] [PubMed]
- Rey, O.; Canon, J.; Krogstad, P. HIV-1 Gag protein associates with F-actin present in microfilaments. Virology 1996, 220, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Dai, R.; Tian, C.J.; Dawson, L.; Gorelick, R.; Yu, X.F. Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. J. Virol. 1999, 73, 2901–2908. [Google Scholar] [PubMed]
- Poole, E.; Strappe, P.; Mok, H.P.; Hicks, R.; Lever, A.M. HIV-1 Gag-RNA interaction occurs at a perinuclear/centrosomal site; analysis by confocal microscopy and FRET. Traffic 2005, 6, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.A.; Puri, A.; Blumenthal, R. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry 2001, 40, 12231–12236. [Google Scholar] [CrossRef] [PubMed]
- Viard, M.; Parolini, I.; Sargiacomo, M.; Fecchi, K.; Ramoni, C.; Ablan, S.; Ruscetti, F.W.; Wang, J.M.; Blumenthal, R. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J. Virol. 2002, 76, 11584–11595. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Hitchen, T.L.; Ootes, L.; McAllery, S.; Wong, A.; Nguyen, K.; McCluskey, A.; Robinson, P.J.; Turville, S.G. HIV infection is influenced by dynamin at 3 independent points in the viral life cycle. Traffic 2017, 18, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Belkina, N.V.; Shaw, S. HIV infection of T cells: Actin-in and actin-out. Sci. Signal. 2009, 2, pe23. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Guo, J.; Wu, Y. The trinity of the cortical actin in the initiation of HIV-1 infection. Retrovirology 2012, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Graziano, F.; Elia, C.; Laudanna, C.; Poli, G.; Alfano, M. Urokinase plasminogen activator inhibits HIV virion release from macrophage-differentiated chronically infected cells via activation of RhoA and PKCepsilon. PLoS ONE 2011, 6, e23674. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A.; de Marco, A.; Oberwinkler, H.; Habermann, A.; Briggs, J.A.; Krausslich, H.G.; Grunewald, K. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 2010, 6, e1001173. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.A.; Koch, P.; Weichsel, J.; Godinez, W.J.; Schwarz, U.; Rohr, K.; Lamb, D.C.; Krausslich, H.G.; Muller, B. Investigating the role of F-actin in human immunodeficiency virus assembly by live-cell microscopy. J. Virol. 2014, 88, 7904–7914. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Mariani-Floderer, C.; Lopez-Huertas, M.R.; Gros, N.; Hamard-Peron, E.; Favard, C.; Ohlmann, T.; Alcami, J.; Muriaux, D. Involvement of the Rac1-IRSp53-Wave2-Arp2/3 Signaling Pathway in HIV-1 Gag Particle Release in CD4 T Cells. J. Virol. 2015, 89, 8162–8181. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Guo, J.; Wu, Y. Novel anti-HIV therapeutics targeting chemokine receptors and actin regulatory pathways. Immunol. Rev. 2013, 256, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, P.; Roy, C.; Martin, M. ERM proteins in cell adhesion and membrane dynamics. Trends Cell Biol. 1999, 9, 187–192. [Google Scholar] [CrossRef]
- Popowicz, G.M.; Schleicher, M.; Noegel, A.A.; Holak, T.A. Filamins: Promiscuous organizers of the cytoskeleton. Trends Biochem. Sci. 2006, 31, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell Adhes. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef]
- Jimenez-Baranda, S.; Gomez-Mouton, C.; Rojas, A.; Martinez-Prats, L.; Mira, E.; Ana Lacalle, R.; Valencia, A.; Dimitrov, D.S.; Viola, A.; Delgado, R.; et al. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007, 9, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; Qian, W.J.; Diamond, D.L.; Liu, T.; Gritsenko, M.A.; Monroe, M.E.; Camp, D.G., 2nd; Smith, R.D.; Katze, M.G. Quantitative analysis of human immunodeficiency virus type 1-infected CD4+ cell proteome: Dysregulated cell cycle progression and nuclear transport coincide with robust virus production. J. Virol. 2007, 81, 7571–7583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jia, X.; Zhang, X.; Sun, J.; Peng, X.; Qi, T.; Ma, F.; Yin, L.; Yao, Y.; Qiu, C.; et al. Proteomic analysis of PBMCs: Characterization of potential HIV-associated proteins. Proteome Sci. 2010, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.; Cylinder, I.; Platt, E.J.; Barklis, E. Analysis of HIV-1 Gag protein interactions via biotin ligase tagging. J. Virol. 2015, 89, 3988–4001. [Google Scholar] [CrossRef] [PubMed]
- Chertova, E.; Chertov, O.; Coren, L.V.; Roser, J.D.; Trubey, C.M.; Bess, J.W., Jr.; Sowder, R.C., 2nd; Barsov, E.; Hood, B.L.; Fisher, R.J.; et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 2006, 80, 9039–9052. [Google Scholar] [CrossRef] [PubMed]
- Ponuwei, G.A. A glimpse of the ERM proteins. J. Biomed. Sci. 2016, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Barrero-Villar, M.; Cabrero, J.R.; Gordon-Alonso, M.; Barroso-Gonzalez, J.; Alvarez-Losada, S.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; Valenzuela-Fernandez, A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Houzet, L.; Yedavalli, V.S.; Jeang, K.T. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 2009, 284, 19463–19473. [Google Scholar] [CrossRef] [PubMed]
- Scheuring, U.J.; Corbeil, J.; Mosier, D.E.; Theofilopoulos, A.N. Early modification of host cell gene expression induced by HIV-1. AIDS 1998, 12, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, G.; Mueller-Kuller, T.; Markovic, S.; Klein, S.A.; Dietrich, U.; Hoelzer, D.; Ottmann, O.G.; Scheuring, U.J. Knockdown of ERM family member moesin in host cells increases HIV type 1 replication. AIDS Res. Hum. Retrovir. 2011, 27, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Woollard, S.M.; Li, H.; Singh, S.; Yu, F.; Kanmogne, G.D. HIV-1 induces cytoskeletal alterations and Rac1 activation during monocyte-blood-brain barrier interactions: Modulatory role of CCR5. Retrovirology 2014, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Frederick, K.M.; Haverland, N.A.; Ciborowski, P.; Belshan, M. Investigation of the HIV-1 matrix interactome during virus replication. Proteom. Clin. Appl. 2016, 10, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.H.; Lambele, M.; Chan, J.; Symeonides, M.; Thali, M. Ezrin is a component of the HIV-1 virological presynapse and contributes to the inhibition of cell-cell fusion. J. Virol. 2014, 88, 7645–7658. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, W.H.; Ezzell, R.M.; Adamson, E.D.; Niggli, V.; Isenberg, G. Vinculin, talin and focal adhesions. J. Muscle Res. Cell Motil. 1996, 17, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bays, J.L.; DeMali, K.A. Vinculin in cell-cell and cell-matrix adhesions. Cell. Mol. Life Sci. 2017, 74, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo Veronese, F.; Arnott, D.; Barnaba, V.; Loftus, D.J.; Sakaguchi, K.; Thompson, C.B.; Salemi, S.; Mastroianni, C.; Sette, A.; Shabanowitz, J.; et al. Autoreactive cytotoxic T lymphocytes in human immunodeficiency virus type 1-infected subjects. J. Exp. Med. 1996, 183, 2509–2516. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Morham, S.G.; Walsh, D.; Naghavi, M.H. Focal adhesion proteins talin-1 and vinculin negatively affect paxillin phosphorylation and limit retroviral infection. J. Mol. Biol. 2011, 410, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Murali, A.; Rajalingam, K. Small Rho GTPases in the control of cell shape and mobility. Cell. Mol. Life Sci. 2014, 71, 1703–1721. [Google Scholar] [CrossRef] [PubMed]
- De Curtis, I.; Meldolesi, J. Cell surface dynamics—How Rho GTPases orchestrate the interplay between the plasma membrane and the cortical cytoskeleton. J. Cell Sci. 2012, 125, 4435–4444. [Google Scholar] [CrossRef] [PubMed]
- Harmon, B.; Ratner, L. Induction of the Galpha(q) signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J. Virol. 2008, 82, 9191–9205. [Google Scholar] [CrossRef] [PubMed]
- Mertens, A.E.; Roovers, R.C.; Collard, J.G. Regulation of Tiam1-Rac signalling. FEBS Lett. 2003, 546, 11–16. [Google Scholar] [CrossRef]
- Vorster, P.J.; Guo, J.; Yoder, A.; Wang, W.; Zheng, Y.; Xu, X.; Yu, D.; Spear, M.; Wu, Y. LIM kinase 1 modulates cortical actin and CXCR4 cycling and is activated by HIV-1 to initiate viral infection. J. Biol. Chem. 2011, 286, 12554–12564. [Google Scholar] [CrossRef] [PubMed]
- Pontow, S.E.; Heyden, N.V.; Wei, S.; Ratner, L. Actin cytoskeletal reorganizations and coreceptor-mediated activation of rac during human immunodeficiency virus-induced cell fusion. J. Virol. 2004, 78, 7138–7147. [Google Scholar] [CrossRef] [PubMed]
- Pontow, S.; Harmon, B.; Campbell, N.; Ratner, L. Antiviral activity of a Rac GEF inhibitor characterized with a sensitive HIV/SIV fusion assay. Virology 2007, 368, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Imamura, J.; Suzuki, Y.; Gonda, K.; Roy, C.N.; Gatanaga, H.; Ohuchi, N.; Higuchi, H. Single particle tracking confirms that multivalent Tat protein transduction domain-induced heparan sulfate proteoglycan cross-linkage activates Rac1 for internalization. J. Biol. Chem. 2011, 286, 10581–10592. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Gu, Y.; Xu, Y.C.; Mitola, S.; Bussolino, F.; Terada, L.S. Human immunodeficiency virus type 1 Tat regulates endothelial cell actin cytoskeletal dynamics through PAK1 activation and oxidant production. J. Virol. 2004, 78, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Ma, Z.; Myers, D.P.; Terada, L.S. HIV-1 Tat activates dual Nox pathways leading to independent activation of ERK and JNK MAP kinases. J. Biol. Chem. 2007, 282, 37412–37419. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.C.; He, J.C.; Wang, Z.H.; Feng, X.; Fukumi-Tominaga, T.; Chen, N.; Xu, J.; Iyengar, R.; Klotman, P.E. HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J. Biol. Chem. 2008, 283, 8173–8182. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Patni, H.; Tandon, P.; Luan, L.; Sharma, B.; Salhan, D.; Saleem, M.A.; Mathieson, P.W.; Malhotra, A.; Husain, M.; et al. Nef interaction with actin compromises human podocyte actin cytoskeletal integrity. Exp. Mol. Pathol. 2013, 94, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, A.; Swigut, T.; Hill, B.; Myers, M.P.; Skowronski, J. HIV-1 Nef binds the DOCK2-ELMO1 complex to activate rac and inhibit lymphocyte chemotaxis. PLoS Biol. 2004, 2, E6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, A.; Das, S.R.; Jameel, S.; George, A.; Bal, V.; Mayor, S.; Rath, S. A two-pronged mechanism for HIV-1 Nef-mediated endocytosis of immune costimulatory molecules CD80 and CD86. Cell Host Microbe 2007, 1, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, M.G.; Mattioli, B.; Spadaro, F.; Straface, E.; Giordani, L.; Ramoni, C.; Malorni, W.; Viora, M. HIV-1 Nef triggers Vav-mediated signaling pathway leading to functional and morphological differentiation of dendritic cells. FASEB J. 2003, 17, 2025–2036. [Google Scholar] [CrossRef] [PubMed]
- Katzav, S. Vav1: A Dr. Jekyll and Mr. Hyde protein—Good for the hematopoietic system, bad for cancer. Oncotarget 2015, 6, 28731–28742. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wu, X.; Plemenitas, A.; Yu, H.; Sawai, E.T.; Abo, A.; Peterlin, B.M. CDC42 and Rac1 are implicated in the activation of the Nef-associated kinase and replication of HIV-1. Curr. Biol. 1996, 6, 1677–1684. [Google Scholar] [CrossRef]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Carrizzo, A.; Forte, M.; Lembo, M.; Formisano, L.; Puca, A.A.; Vecchione, C. Rac-1 as a new therapeutic target in cerebro- and cardio-vascular diseases. Curr. Drug Targets 2014, 15, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Feng, X.; Shi, M.; Cai, Q.; Yu, Y.; Zhu, Z.; Zhang, J. Rac1 is correlated with aggressiveness and a potential therapeutic target for gastric cancer. Int. J. Oncol. 2015, 46, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.; Malliri, A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 2017, 8, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Swaine, T.; Dittmar, M.T. CDC42 Use in Viral Cell Entry Processes by RNA Viruses. Viruses 2015, 7, 6526–6536. [Google Scholar] [CrossRef] [PubMed]
- Lucera, M.B.; Fleissner, Z.; Tabler, C.O.; Schlatzer, D.M.; Troyer, Z.; Tilton, J.C. HIV signaling through CD4 and CCR5 activates Rho family GTPases that are required for optimal infection of primary CD4+ T cells. Retrovirology 2017, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Len, A.C.L.; Starling, S.; Shivkumar, M.; Jolly, C. HIV-1 Activates T Cell Signaling Independently of Antigen to Drive Viral Spread. Cell Rep. 2017, 18, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Svajger, U.; Anderluh, M.; Jeras, M.; Obermajer, N. C-type lectin DC-SIGN: An adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell. Signal. 2010, 22, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Luo, W.; Geyer, M.; Alberts, A.S.; Peterlin, B.M. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol. Cell 1999, 3, 729–739. [Google Scholar] [CrossRef]
- Chou, R.; Dana, T.; Blazina, I.; Daeges, M.; Jeanne, T.L. Statins for Prevention of Cardiovascular Disease in Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2016, 316, 2008–2024. [Google Scholar] [CrossRef] [PubMed]
- Del Real, G.; Jimenez-Baranda, S.; Mira, E.; Lacalle, R.A.; Lucas, P.; Gomez-Mouton, C.; Alegret, M.; Pena, J.M.; Rodriguez-Zapata, M.; Alvarez-Mon, M.; et al. Statins inhibit HIV-1 infection by down-regulating Rho activity. J. Exp. Med. 2004, 200, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Eckard, A.R.; Meissner, E.G.; Singh, I.; McComsey, G.A. Cardiovascular Disease, Statins, and HIV. J. Infect. Dis. 2016, 214 (Suppl. S2), S83–S92. [Google Scholar] [CrossRef] [PubMed]
- Skau, C.T.; Waterman, C.M. Specification of Architecture and Function of Actin Structures by Actin Nucleation Factors. Annu. Rev. Biophys. 2015, 44, 285–310. [Google Scholar] [CrossRef] [PubMed]
- Chesarone, M.A.; Goode, B.L. Actin nucleation and elongation factors: Mechanisms and interplay. Curr. Opin. Cell Biol. 2009, 21, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Mechanobio Website. Available online: https://www.mechanobio.info/topics/cytoskeleton-dynamics/actin-nucleation/ (accessed on 11 November 2017).
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Komano, J.; Miyauchi, K.; Matsuda, Z.; Yamamoto, N. Inhibiting the Arp2/3 complex limits infection of both intracellular mature vaccinia virus and primate lentiviruses. Mol. Biol. Cell 2004, 15, 5197–5207. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Prasad, V.R. Analysis of 2-LTR circle junctions of viral DNA in infected cells. Methods Mol. Biol. 2009, 485, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Menager, M.M.; Littman, D.R. Actin Dynamics Regulates Dendritic Cell-Mediated Transfer of HIV-1 to T Cells. Cell 2016, 164, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.; Copeland, J.W.; Brumell, J.H. Bacterial subversion of host cytoskeletal machinery: Hijacking formins and the Arp2/3 complex. BioEssays 2014, 36, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e29513. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, S.; Schultz, J.; Grosshans, J. Formin’ cellular structures: Physiological roles of Diaphanous (Dia) in actin dynamics. Commun. Integr. Biol. 2013, 6, e27634. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, F.; Gundersen, G.G. Formins and microtubules. Biochim. Biophys. Acta 2010, 1803, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Pring, M.; Evangelista, M.; Boone, C.; Yang, C.; Zigmond, S.H. Mechanism of formin-induced nucleation of actin filaments. Biochemistry 2003, 42, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Delaney, M.K.; Malikov, V.; Chai, Q.; Zhao, G.; Naghavi, M.H. Distinct functions of diaphanous-related formins regulate HIV-1 uncoating and transport. Proc. Natl. Acad. Sci. USA 2017, 114, E6932–E6941. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Lin, C.; Hu, F.; Wang, F.; Zhu, L.; Yao, X.; Wang, Y.; Zhao, Y. APC polymorphisms and the risk of colorectal neoplasia: A HuGE review and meta-analysis. Am. J. Epidemiol. 2013, 177, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Firat-Karalar, E.N.; Welch, M.D. New mechanisms and functions of actin nucleation. Curr. Opin. Cell Biol. 2011, 23, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Bartolini, F.; Deaconescu, A.M.; Moseley, J.B.; Dogic, Z.; Grigorieff, N.; Gundersen, G.G.; Goode, B.L. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J. Cell Biol. 2010, 189, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Breitsprecher, D.; Jaiswal, R.; Bombardier, J.P.; Gould, C.J.; Gelles, J.; Goode, B.L. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science 2012, 336, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Taketo, M.M. Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. J. Cell Sci. 2007, 120, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.A.; Bouguenina, H.; Eskin, J.A.; Jaiswal, R.; Badache, A.; Goode, B.L. Adenomatous polyposis coli nucleates actin assembly to drive cell migration and microtubule-induced focal adhesion turnover. J. Cell Biol. 2017, 216, 2859–2875. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, S.; Carlier, M.F. Enhanced Depolymerization of Actin Filaments by ADF/Cofilin and Monomer Funneling by Capping Protein Cooperate to Accelerate Barbed-End Growth. Curr. Biol. 2017, 27, 1990–1998.e5. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, O.; Vicente-Manzanares, M.; Urzainqui, A.; Yanez-Mo, M.; Sanchez-Madrid, F. Interactive protrusive structures during leukocyte adhesion and transendothelial migration. Front. Biosci. 2004, 9, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Ono, S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int. Rev. Cytol. 2007, 258, 1–82. [Google Scholar] [CrossRef] [PubMed]
- Kanellos, G.; Frame, M.C. Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Bamburg, J.R. ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Wioland, H.; Guichard, B.; Senju, Y.; Myram, S.; Lappalainen, P.; Jegou, A.; Romet-Lemonne, G. ADF/Cofilin Accelerates Actin Dynamics by Severing Filaments and Promoting Their Depolymerization at Both Ends. Curr. Biol. 2017, 27, 1956–1967.e7. [Google Scholar] [CrossRef] [PubMed]
- Yoder, A.; Yu, D.; Dong, L.; Iyer, S.R.; Xu, X.; Kelly, J.; Liu, J.; Wang, W.; Vorster, P.J.; Agulto, L.; et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 2008, 134, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, W.; Yu, D.; Wu, Y. Spinoculation triggers dynamic actin and cofilin activity that facilitates HIV-1 infection of transformed and resting CD4 T cells. J. Virol. 2011, 85, 9824–9833. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yoder, A.; Yu, D.; Wang, W.; Liu, J.; Barrett, T.; Wheeler, D.; Schlauch, K. Cofilin activation in peripheral CD4 T cells of HIV-1 infected patients: A pilot study. Retrovirology 2008, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Q.; Yamamoto, M.; Mejillano, M.; Yin, H.L. Gelsolin, a multifunctional actin regulatory protein. J. Biol. Chem. 1999, 274, 33179–33182. [Google Scholar] [CrossRef] [PubMed]
- Boukli, N.M.; Shetty, V.; Cubano, L.; Ricaurte, M.; Coelho-Dos-Reis, J.; Nickens, Z.; Shah, P.; Talal, A.H.; Philip, R.; Jain, P. Unique and differential protein signatures within the mononuclear cells of HIV-1 and HCV mono-infected and co-infected patients. Clin. Proteom. 2012, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.K.; Peddada, N.; Jha, P.K.; Mishra, A.; Pandey, K.; Das, V.N.; Ashish; Das, P. Plasma Gelsolin Level in HIV-1-Infected Patients: An Indicator of Disease Severity. AIDS Res. Hum. Retrovir. 2017, 33, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef] [PubMed]
- Coiras, M.; Camafeita, E.; Urena, T.; Lopez, J.A.; Caballero, F.; Fernandez, B.; Lopez-Huertas, M.R.; Perez-Olmeda, M.; Alcami, J. Modifications in the human T cell proteome induced by intracellular HIV-1 Tat protein expression. Proteomics 2006, 6 (Suppl. S1), S63–S73. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Exposito, L.; Ziglio, S.; Barroso-Gonzalez, J.; de Armas-Rillo, L.; Valera, M.S.; Zipeto, D.; Machado, J.D.; Valenzuela-Fernandez, A. Gelsolin activity controls efficient early HIV-1 infection. Retrovirology 2013, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Numazaki, M.; Takeuchi, K.; Uchibori, Y.; Ando-Akatsuka, Y.; Tominaga, M.; Tominaga, T. DIP (mDia interacting protein) is a key molecule regulating Rho and Rac in a Src-dependent manner. EMBO J. 2004, 23, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, H.; Solski, P.A.; Hart, M.J.; Der, C.J.; Su, L. Modulation of HIV-1 replication by a novel RhoA effector activity. J. Immunol. 2000, 164, 5369–5374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, L.; Kao, S.; Whitehead, I.P.; Hart, M.J.; Liu, B.; Duus, K.; Burridge, K.; Der, C.J.; Su, L. Functional interaction between the cytoplasmic leucine-zipper domain of HIV-1 gp41 and p115-RhoGEF. Curr. Biol. 1999, 9, 1271–1274. [Google Scholar] [CrossRef]
- Zhong, Y.; Hennig, B.; Toborek, M. Intact lipid rafts regulate HIV-1 Tat protein-induced activation of the Rho signaling and upregulation of P-glycoprotein in brain endothelial cells. J. Cereb. Blood Flow Metab. 2010, 30, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Colberg-Poley, A.M.; Das, J.R.; Li, J.; Zhang, A.; Tang, P.; Jerebtsova, M.; Gutkind, J.S.; Ray, P.E. The basic domain of HIV-tat transactivating protein is essential for its targeting to lipid rafts and regulating fibroblast growth factor-2 signaling in podocytes isolated from children with HIV-1-associated nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1800–1813. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Huertas, M.R.; Callejas, S.; Abia, D.; Mateos, E.; Dopazo, A.; Alcami, J.; Coiras, M. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucleic Acids Res. 2010, 38, 3287–3307. [Google Scholar] [CrossRef] [PubMed]
- Ishaq, M.; Lin, B.R.; Bosche, M.; Zheng, X.; Yang, J.; Huang, D.; Lempicki, R.A.; Aguilera-Gutierrez, A.; Natarajan, V. LIM kinase 1—Dependent cofilin 1 pathway and actin dynamics mediate nuclear retinoid receptor function in T lymphocytes. BMC Mol. Biol. 2011, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Reichman-Fried, M.; Abraham, L.; Pan, X.; Giese, S.I.; Hannemann, S.; Goulimari, P.; Raz, E.; Grosse, R.; Fackler, O.T. HIV-1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe 2009, 6, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Abraham, L.; Rudolph, J.M.; Fackler, O.T. Lentiviral Nef proteins utilize PAK2-mediated deregulation of cofilin as a general strategy to interfere with actin remodeling. J. Virol. 2010, 84, 3935–3948. [Google Scholar] [CrossRef] [PubMed]
- Verollet, C.; Souriant, S.; Bonnaud, E.; Jolicoeur, P.; Raynaud-Messina, B.; Kinnaer, C.; Fourquaux, I.; Imle, A.; Benichou, S.; Fackler, O.T.; et al. HIV-1 reprograms the migration of macrophages. Blood 2015, 125, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Rauch, S.; Michel, N.; Hannemann, S.; Lehmann, M.J.; Keppler, O.T.; Fackler, O.T. The HIV-1 pathogenicity factor Nef interferes with maturation of stimulatory T-lymphocyte contacts by modulation of N-Wasp activity. J. Biol. Chem. 2006, 281, 19618–19630. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K. Interactions of the HIV/SIV pathogenicity factor Nef with SH3 domain-containing host cell proteins. Curr. HIV Res. 2011, 9, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Pulkkinen, K.; Saksela, K.; Fackler, O.T. Human immunodeficiency virus type 1 Nef recruits the guanine exchange factor Vav1 via an unexpected interface into plasma membrane microdomains for association with p21-activated kinase 2 activity. J. Virol. 2008, 82, 2918–2929. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, E.; Giese, S.I.; Gasteier, J.E.; Muranyi, W.; Fackler, O.T. Human immunodeficiency virus type 1 Nef activates p21-activated kinase via recruitment into lipid rafts. J. Virol. 2004, 78, 4085–4097. [Google Scholar] [CrossRef] [PubMed]
- Sawai, E.T.; Baur, A.; Struble, H.; Peterlin, B.M.; Levy, J.A.; Cheng-Mayer, C. Human immunodeficiency virus type 1 Nef associates with a cellular serine kinase in T lymphocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Reeder, M.K.; Serebriiskii, I.G.; Golemis, E.A.; Chernoff, J. Analysis of small GTPase signaling pathways using p21-activated kinase mutants that selectively couple to Cdc42. J. Biol. Chem. 2001, 276, 40606–40613. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Lu, X.; Frost, J.A.; Geyer, M.; Jiang, B.; Luo, W.; Abo, A.; Alberts, A.S.; Peterlin, B.M. p21-activated kinase 1 plays a critical role in cellular activation by Nef. Mol. Cell. Biol. 2000, 20, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Manninen, A.; Mann, D.A.; Harris, M.; Saksela, K. Identification of the Nef-associated kinase as p21-activated kinase 2. Curr. Biol. 1999, 9, 1407–1410. [Google Scholar] [CrossRef]
- Nunn, M.F.; Marsh, J.W. Human immunodeficiency virus type 1 Nef associates with a member of the p21-activated kinase family. J. Virol. 1996, 70, 6157–6161. [Google Scholar] [PubMed]
- Cullen, B.R. HIV-1: Is Nef a PAK animal? Curr. Biol. 1996, 6, 1557–1559. [Google Scholar] [CrossRef]
- Wiskerchen, M.; Cheng-Mayer, C. HIV-1 Nef association with cellular serine kinase correlates with enhanced virion infectivity and efficient proviral DNA synthesis. Virology 1996, 224, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Kouwenhoven, A.; Minassian, V.D.; Marsh, J.W. HIV-1 Nef mediates Pak phosphorylation of Mek1 serine298 and elicits an active phospho-state of Pak2. Curr. HIV Res. 2013, 11, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, T.; Zheng, Y.H.; Mandic, R.; Peterlin, B.M. Interaction between Nef and phosphatidylinositol-3-kinase leads to activation of p21-activated kinase and increased production of HIV. Virology 2002, 294, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Pulkkinen, K.; Renkema, G.H.; Kirchhoff, F.; Saksela, K. Nef associates with p21-activated kinase 2 in a p21-GTPase-dependent dynamic activation complex within lipid rafts. J. Virol. 2004, 78, 12773–12780. [Google Scholar] [CrossRef] [PubMed]
- Sawai, E.T.; Khan, I.H.; Montbriand, P.M.; Peterlin, B.M.; Cheng-Mayer, C.; Luciw, P.A. Activation of PAK by HIV and SIV Nef: Importance for AIDS in rhesus macaques. Curr. Biol. 1996, 6, 1519–1527. [Google Scholar] [CrossRef]
- Khan, I.H.; Sawai, E.T.; Antonio, E.; Weber, C.J.; Mandell, C.P.; Montbriand, P.; Luciw, P.A. Role of the SH3-ligand domain of simian immunodeficiency virus Nef in interaction with Nef-associated kinase and simian AIDS in rhesus macaques. J. Virol. 1998, 72, 5820–5830. [Google Scholar] [PubMed]
- Imle, A.; Abraham, L.; Tsopoulidis, N.; Hoflack, B.; Saksela, K.; Fackler, O.T. Association with PAK2 Enables Functional Interactions of Lentiviral Nef Proteins with the Exocyst Complex. mBio 2015, 6, e01309-15. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Manninen, A.; Saksela, K. Human immunodeficiency virus type 1 Nef selectively associates with a catalytically active subpopulation of p21-activated kinase 2 (PAK2) independently of PAK2 binding to Nck or beta-PIX. J. Virol. 2001, 75, 2154–2160. [Google Scholar] [CrossRef] [PubMed]
- Moarefi, I.; LaFevre-Bernt, M.; Sicheri, F.; Huse, M.; Lee, C.H.; Kuriyan, J.; Miller, W.T. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 1997, 385, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Guiet, R.; Poincloux, R.; Castandet, J.; Marois, L.; Labrousse, A.; Le Cabec, V.; Maridonneau-Parini, I. Hematopoietic cell kinase (Hck) isoforms and phagocyte duties—From signaling and actin reorganization to migration and phagocytosis. Eur. J. Cell Biol. 2008, 87, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Saksela, K.; Cheng, G.; Baltimore, D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 1995, 14, 484–491. [Google Scholar] [PubMed]
- Lee, C.H.; Leung, B.; Lemmon, M.A.; Zheng, J.; Cowburn, D.; Kuriyan, J.; Saksela, K. A single amino acid in the SH3 domain of Hck determines its high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 1995, 14, 5006–5015. [Google Scholar] [PubMed]
- Lerner, E.C.; Smithgall, T.E. SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat. Struct. Biol. 2002, 9, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, J.J.; Tarafdar, S.; Yeh, J.I.; Smithgall, T.E. Interaction with the Src homology (SH3-SH2) region of the Src-family kinase Hck structures the HIV-1 Nef dimer for kinase activation and effector recruitment. J. Biol. Chem. 2014, 289, 28539–28553. [Google Scholar] [CrossRef] [PubMed]
- Komuro, I.; Yokota, Y.; Yasuda, S.; Iwamoto, A.; Kagawa, K.S. CSF-induced and HIV-1-mediated distinct regulation of Hck and C/EBPbeta represent a heterogeneous susceptibility of monocyte-derived macrophages to M-tropic HIV-1 infection. J. Exp. Med. 2003, 198, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Trible, R.P.; Narute, P.; Emert-Sedlak, L.A.; Alvarado, J.J.; Atkins, K.; Thomas, L.; Kodama, T.; Yanamala, N.; Korotchenko, V.; Day, B.W.; et al. Discovery of a diaminoquinoxaline benzenesulfonamide antagonist of HIV-1 Nef function using a yeast-based phenotypic screen. Retrovirology 2013, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Cornall, A.; Mak, J.; Greenway, A.; Tachedjian, G. HIV-1 infection of T cells and macrophages are differentially modulated by virion-associated Hck: A Nef-dependent phenomenon. Viruses 2013, 5, 2235–2252. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, K.; Kojima, A.; Kurata, T.; Ikuta, K.; Akari, H.; Koyama, A.H.; Kawamura, M.; Inubushi, R.; Shimano, R.; Adachi, A. Enhancement of human immunodeficiency virus type 1 infectivity by Nef is producer cell-dependent. J. Gen. Virol. 1998, 79 Pt 10, 2447–2453. [Google Scholar] [CrossRef] [PubMed]
- Hanna, Z.; Weng, X.; Kay, D.G.; Poudrier, J.; Lowell, C.; Jolicoeur, P. The pathogenicity of human immunodeficiency virus (HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-binding domain, and disease development is delayed in the absence of Hck. J. Virol. 2001, 75, 9378–9392. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, F.; Schenone, S.; Brullo, C.; Desogus, A.; Botta, L.; Tintori, C. Hck inhibitors as potential therapeutic agents in cancer and HIV infection. Curr. Med. Chem. 2015, 22, 1540–1564. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Shankar, E.M.; Che, K.F.; Saeidi, A.; Ellegard, R.; Barathan, M.; Velu, V.; Kamarulzaman, A. Molecular signatures of T-cell inhibition in HIV-1 infection. Retrovirology 2013, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Choe, E.Y.; Schoenberger, E.S.; Groopman, J.E.; Park, I.W. HIV Nef inhibits T cell migration. J. Biol. Chem. 2002, 277, 46079–46084. [Google Scholar] [CrossRef] [PubMed]
- Park, I.W.; He, J.J. HIV-1 Nef-mediated inhibition of T cell migration and its molecular determinants. J. Leukoc. Biol. 2009, 86, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.; Rudnicka, D.; Hasan, M.; Aulner, N.; Porrot, F.; Machu, C.; Renaud, O.; Prevost, M.C.; Hivroz, C.; Schwartz, O.; et al. HIV-1 Nef inhibits ruffles, induces filopodia, and modulates migration of infected lymphocytes. J. Virol. 2010, 84, 2282–2293. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Imle, A.; Coelho, F.M.; Hons, M.; Gorina, R.; Lyck, R.; Stein, J.V.; Fackler, O.T. HIV-1 Nef interferes with T-lymphocyte circulation through confined environments in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 18541–18546. [Google Scholar] [CrossRef] [PubMed]
- Cernuda-Morollon, E.; Millan, J.; Shipman, M.; Marelli-Berg, F.M.; Ridley, A.J. Rac activation by the T-cell receptor inhibits T cell migration. PLoS ONE 2010, 5, e12393. [Google Scholar] [CrossRef] [PubMed]
- Vene, R.; Benelli, R.; Noonan, D.M.; Albini, A. HIV-Tat dependent chemotaxis and invasion, key aspects of tat mediated pathogenesis. Clin. Exp. Metastasis 2000, 18, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Alcover, A.; Schwartz, O. Modulation of the immunological synapse: A key to HIV-1 pathogenesis? Nat. Rev. Immunol. 2007, 7, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Thoulouze, M.I.; Sol-Foulon, N.; Blanchet, F.; Dautry-Varsat, A.; Schwartz, O.; Alcover, A. Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity 2006, 24, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Abraham, L.; Fackler, O.T. HIV-1 Nef: A multifaceted modulator of T cell receptor signaling. Cell Commun. Signal. 2012, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collette, Y.; Dutartre, H.; Benziane, A.; Ramos, M.; Benarous, R.; Harris, M.; Olive, D. Physical and functional interaction of Nef with Lck. HIV-1 Nef-induced T-cell signaling defects. J. Biol. Chem. 1996, 271, 6333–6341. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Rauch, S.; Fackler, O.T. HIV-1 Nef employs two distinct mechanisms to modulate Lck subcellular localization and TCR induced actin remodeling. PLoS ONE 2007, 2, e1212. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Rudolph, J.M.; Abraham, L.; Habermann, A.; Haller, C.; Krijnse-Locker, J.; Fackler, O.T. HIV-1 Nef compensates for disorganization of the immunological synapse by inducing trans-Golgi network-associated Lck signaling. Blood 2012, 119, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Curado, S.; Mayya, V.; Dustin, M.L. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim. Biophys. Acta 2014, 1838, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.M.; Eickel, N.; Haller, C.; Schindler, M.; Fackler, O.T. Inhibition of t-cell receptor-induced actin remodeling and relocalization of lck are evolutionarily conserved activities of lentiviral nef proteins. J. Virol. 2009, 83, 11528–11539. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Tibroni, N.; Rudolph, J.M.; Grosse, R.; Fackler, O.T. Nef does not inhibit F-actin remodelling and HIV-1 cell-cell transmission at the T lymphocyte virological synapse. Eur. J. Cell Biol. 2011, 90, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Hioe, C.E.; Tuen, M.; Vasiliver-Shamis, G.; Alvarez, Y.; Prins, K.C.; Banerjee, S.; Nadas, A.; Cho, M.W.; Dustin, M.L.; Kachlany, S.C. HIV envelope gp120 activates LFA-1 on CD4 T-lymphocytes and increases cell susceptibility to LFA-1-targeting leukotoxin (LtxA). PLoS ONE 2011, 6, e23202. [Google Scholar] [CrossRef] [PubMed]
- Arthos, J.; Cicala, C.; Martinelli, E.; Macleod, K.; van Ryk, D.; Wei, D.; Xiao, Z.; Veenstra, T.D.; Conrad, T.P.; Lempicki, R.A.; et al. HIV-1 envelope protein binds to and signals through integrin α4β7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 2008, 9, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Mitar, I.; Sattentau, Q.J. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 2007, 81, 13916–13921. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, D.; Feldmann, J.; Porrot, F.; Wietgrefe, S.; Guadagnini, S.; Prevost, M.C.; Estaquier, J.; Haase, A.T.; Sol-Foulon, N.; Schwartz, O. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J. Virol. 2009, 83, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Welsch, S.; Michor, S.; Sattentau, Q.J. The regulated secretory pathway in CD4(+) T cells contributes to human immunodeficiency virus type-1 cell-to-cell spread at the virological synapse. PLoS Pathog. 2011, 7, e1002226. [Google Scholar] [CrossRef] [PubMed]
- Groppelli, E.; Starling, S.; Jolly, C. Contact-induced mitochondrial polarization supports HIV-1 virological synapse formation. J. Virol. 2015, 89, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Starling, S.; Jolly, C. LFA-1 Engagement Triggers T Cell Polarization at the HIV-1 Virological Synapse. J. Virol. 2016, 90, 9841–9854. [Google Scholar] [CrossRef] [PubMed]
- Sol-Foulon, N.; Sourisseau, M.; Porrot, F.; Thoulouze, M.I.; Trouillet, C.; Nobile, C.; Blanchet, F.; di Bartolo, V.; Noraz, N.; Taylor, N.; et al. ZAP-70 kinase regulates HIV cell-to-cell spread and virological synapse formation. EMBO J. 2007, 26, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hubner, W.; Spinelli, M.A.; Chen, B.K. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 2007, 81, 12582–12595. [Google Scholar] [CrossRef] [PubMed]
- Pilhofer, M.; Aistleitner, K.; Ladinsky, M.S.; Konig, L.; Horn, M.; Jensen, G.J. Architecture and host interface of environmental chlamydiae revealed by electron cryotomography. Environ. Microbiol. 2014, 16, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Do, T.; Murphy, G.; Earl, L.A.; Del Prete, G.Q.; Grandinetti, G.; Li, G.H.; Estes, J.D.; Rao, P.; Trubey, C.M.; Thomas, J.; et al. Three-dimensional imaging of HIV-1 virological synapses reveals membrane architectures involved in virus transmission. J. Virol. 2014, 88, 10327–10339. [Google Scholar] [CrossRef] [PubMed]
- Debaisieux, S.; Lachambre, S.; Gross, A.; Mettling, C.; Besteiro, S.; Yezid, H.; Henaff, D.; Chopard, C.; Mesnard, J.M.; Beaumelle, B. HIV-1 Tat inhibits phagocytosis by preventing the recruitment of Cdc42 to the phagocytic cup. Nat. Commun. 2015, 6, 6211. [Google Scholar] [CrossRef] [PubMed]
- Raborn, E.S.; Jamerson, M.; Marciano-Cabral, F.; Cabral, G.A. Cannabinoid inhibits HIV-1 Tat-stimulated adhesion of human monocyte-like cells to extracellular matrix proteins. Life Sci. 2014, 104, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lafrenie, R.M.; Wahl, L.M.; Epstein, J.S.; Hewlett, I.K.; Yamada, K.M.; Dhawan, S. HIV-1-Tat modulates the function of monocytes and alters their interactions with microvessel endothelial cells. A mechanism of HIV pathogenesis. J. Immunol. 1996, 156, 1638–1645. [Google Scholar] [PubMed]
- Matzen, K.; Dirkx, A.E.; oude Egbrink, M.G.; Speth, C.; Gotte, M.; Ascherl, G.; Grimm, T.; Griffioen, A.W.; Sturzl, M. HIV-1 Tat increases the adhesion of monocytes and T-cells to the endothelium in vitro and in vivo: Implications for AIDS-associated vasculopathy. Virus Res. 2004, 104, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Lafrenie, R.M.; Wahl, L.M.; Epstein, J.S.; Hewlett, I.K.; Yamada, K.M.; Dhawan, S. HIV-1-Tat protein promotes chemotaxis and invasive behavior by monocytes. J. Immunol. 1996, 157, 974–977. [Google Scholar] [PubMed]
- Pieper, G.M.; Olds, C.L.; Bub, J.D.; Lindholm, P.F. Transfection of human endothelial cells with HIV-1 tat gene activates NF-kappa B and enhances monocyte adhesion. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2315–H2321. [Google Scholar] [CrossRef] [PubMed]
- De Paulis, A.; de Palma, R.; Di Gioia, L.; Carfora, M.; Prevete, N.; Tosi, G.; Accolla, R.S.; Marone, G. Tat protein is an HIV-1-encoded beta-chemokine homolog that promotes migration and up-regulates CCR3 expression on human Fc epsilon RI+ cells. J. Immunol. 2000, 165, 7171–7179. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.H.; Hearps, A.C.; Martin, G.E.; Williams, K.C.; Crowe, S.M. The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. Aids 2014, 28, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Kuzontkoski, P.M.; Shrivastava, A.; Zhu, W.; Li, D.Y.; Groopman, J.E. Slit2N/Robo1 inhibit HIV-gp120-induced migration and podosome formation in immature dendritic cells by sequestering LSP1 and WASp. PLoS ONE 2012, 7, e48854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, A.R.; Prasad, A.; Bradley, R.R.; Deol, Y.S.; Nagaraja, T.; Ren, X.; Terwilliger, E.F.; Ganju, R.K. HIV-1 gp120-induced migration of dendritic cells is regulated by a novel kinase cascade involving Pyk2, p38 MAP kinase, and LSP1. Blood 2009, 114, 3588–3600. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ospina Stella, A.; Turville, S. All-Round Manipulation of the Actin Cytoskeleton by HIV. Viruses 2018, 10, 63. https://doi.org/10.3390/v10020063
Ospina Stella A, Turville S. All-Round Manipulation of the Actin Cytoskeleton by HIV. Viruses. 2018; 10(2):63. https://doi.org/10.3390/v10020063
Chicago/Turabian StyleOspina Stella, Alberto, and Stuart Turville. 2018. "All-Round Manipulation of the Actin Cytoskeleton by HIV" Viruses 10, no. 2: 63. https://doi.org/10.3390/v10020063
APA StyleOspina Stella, A., & Turville, S. (2018). All-Round Manipulation of the Actin Cytoskeleton by HIV. Viruses, 10(2), 63. https://doi.org/10.3390/v10020063