Cytoskeletons in the Closet—Subversion in Alphaherpesvirus Infections

 ,
,  and
and
Abstract
:1. Introduction
2. Herpesviruses
2.1. Introduction
2.2. General Properties of Alphaherpesvirinae
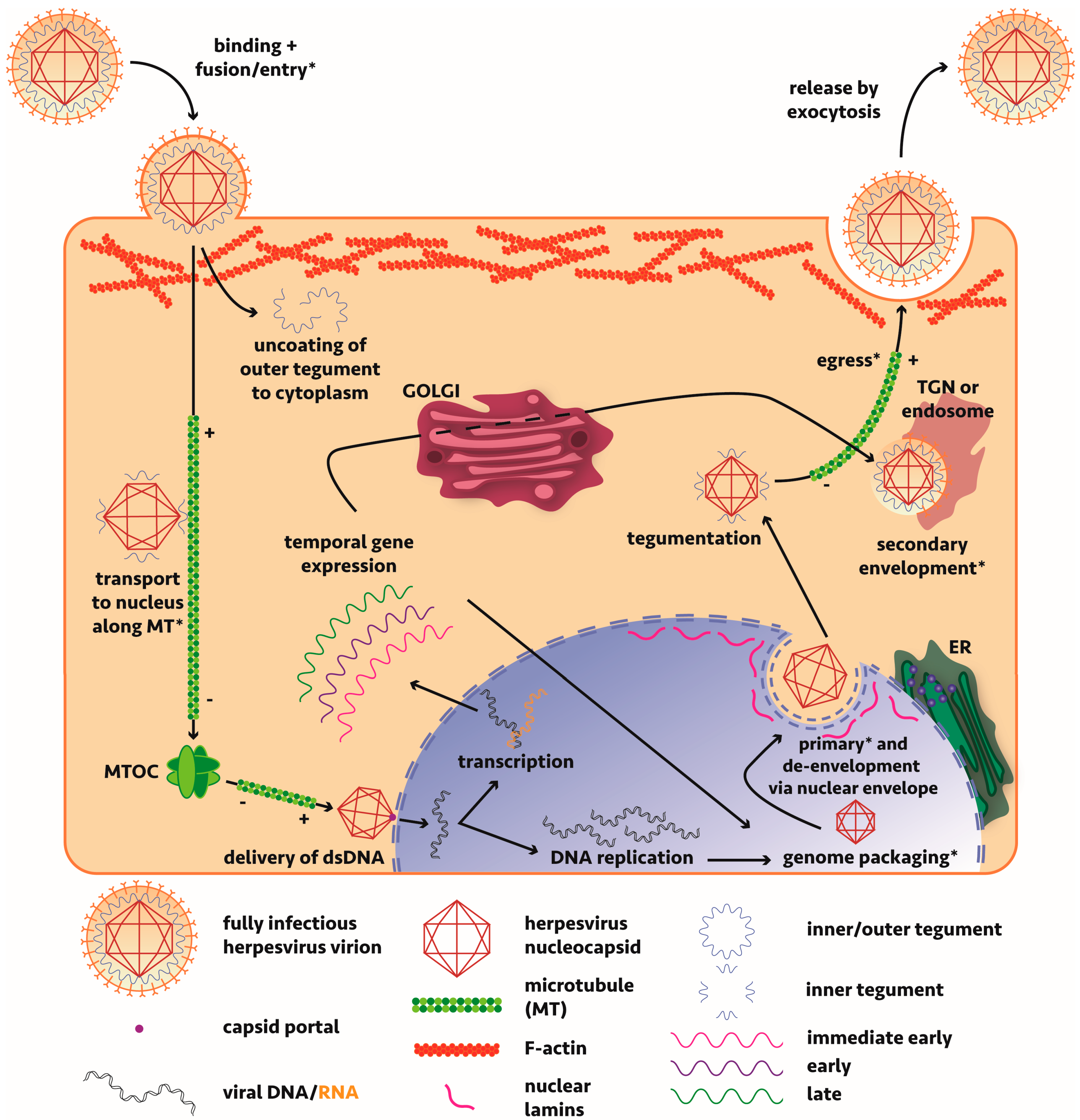
2.3. Maturation Cycle of Alphaherpesvirinae in Non-Neuronal Cells
3. Filamentous Actin
3.1. Introduction
3.2. Actin Remodelling by Alphaherpesviruses
3.3. Alphaherpesvirus Exploitation of Actin-Based Transport and Myosin
3.4. Actin Remodelling and Myosin Motor Exploitation by other Herpesviruses
3.5. Future Work for Alphaherpesvirus-Actin Interaction Studies
4. Microtubules
4.1. Introduction
4.2. Microtubule Remodelling by Alphaherpesviruses
4.3. Alphaherpesvirus Exploitation of Microtubule-Based Transport Motors
4.4. Microtubule Remodelling by Other Herpesviruses
4.5. Future Work for Alphaherpesvirus-Microtubule Interaction Studies
5. Intermediate Filaments
5.1. Introduction
5.2. Intermediate Filament Remodelling by Alphaherpesviruses
5.3. Future Work for Alphaherpesvirus-Intermediate Filament Interaction Studies
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. What We Know and Do Not Know About Actin. Handb. Exp. Pharmacol. 2017, 235, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Bar, H.; Kreplak, L.; Strelkov, S.V.; Aebi, U. Intermediate filaments: From cell architecture to nanomechanics. Nat. Rev. Mol. Cell Biol. 2007, 8, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, W.H. Intermediate Filaments and Cellular Mechanics. Cell Biol. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Marzook, N.B.; Newsome, T.P. Viruses That Exploit Actin-Based Motility for Their Replication and Spread. Handb. Exp. Pharmacol. 2017, 235, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Colonne, P.M.; Winchell, C.G.; Voth, D.E. Hijacking Host Cell Highways: Manipulation of the Host Actin Cytoskeleton by Obligate Intracellular Bacterial Pathogens. Front. Cell Infect. Microbiol. 2016, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Lamason, R.L.; Welch, M.D. Actin-based motility and cell-to-cell spread of bacterial pathogens. Curr. Opin. Microbiol. 2017, 35, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.H.; Walsh, D. Microtubule Regulation and Function during Virus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, G.K.; Splitter, G.A. Modulation of host microtubule dynamics by pathogenic bacteria. Biomol. Concepts 2012, 3, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822. [Google Scholar]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Croen, K.D.; Ostrove, J.M.; Dragovic, L.J.; Straus, S.E. Patterns of gene expression and sites of latency in human nerve ganglia are different for varicella-zoster and herpes simplex viruses. Proc. Natl. Acad. Sci. USA 1988, 85, 9773–9777. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, I.; Gottlieb, J.; Meier, A.; Sohr, D.; Ruhparwar, A.; Henke-Gendo, C.; Gastmeier, P.; Welte, T.; Schulz, T.F.; Mattner, F. Clinical relevance of and risk factors for HSV-related tracheobronchitis or pneumonia: Results of an outbreak investigation. Crit. Care 2007, 11, R119. [Google Scholar] [CrossRef] [PubMed]
- Mori, I.; Nishiyama, Y. Herpes simplex virus and varicella-zoster virus: Why do these human alphaherpesviruses behave so differently from one another? Rev. Med. Virol. 2005, 15, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Genomic structure and organization of varicella-zoster virus. Contrib. Microbiol. 1999, 3, 10–20. [Google Scholar] [PubMed]
- Nishiyama, Y. Herpes simplex virus gene products: The accessories reflect her lifestyle well. Rev. Med. Virol. 2004, 14, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Hertel, L. Herpesviruses and intermediate filaments: Close encounters with the third type. Viruses 2011, 3, 1015–1040. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Moffat, J.F.; Sommer, M.; Oliver, S.; Che, X.; Vleck, S.; Zerboni, L.; Ku, C.C. Varicella-zoster virus T cell tropism and the pathogenesis of skin infection. Curr. Top. Microbiol. Immunol. 2010, 342, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Sacks, S.L.; Griffiths, P.D.; Corey, L.; Cohen, C.; Cunningham, A.; Dusheiko, G.M.; Self, S.; Spruance, S.; Stanberry, L.R.; Wald, A.; et al. HSV shedding. Antivir. Res. 2004, 63 (Suppl. S1), S19–S26. [Google Scholar] [CrossRef] [PubMed]
- Pergam, S.A.; Limaye, A.P.; Practice, A.S.T.I.D.C.o. Varicella zoster virus (VZV) in solid organ transplant recipients. Am. J. Transplant. 2009, 9 (Suppl. S4), S108–S115. [Google Scholar] [CrossRef] [PubMed]
- Sodeik, B. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 2000, 8, 465–472. [Google Scholar] [CrossRef]
- Kukhanova, M.K.; Korovina, A.N.; Kochetkov, S.N. Human herpes simplex virus: Life cycle and development of inhibitors. Biochemistry 2014, 79, 1635–1652. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.C.; Feng, H.; Lin, Y.C.; Guo, X.R. New strategies against drug resistance to herpes simplex virus. Int. J. Oral Sci. 2016, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, N.A.; Rizos, H.; Diefenbach, R.J. Oncolytic virotherapy using herpes simplex virus: How far have we come? Oncolytic Virother. 2015, 4, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Goins, W.F.; Hall, B.; Cohen, J.B.; Glorioso, J.C. Retargeting of herpes simplex virus (HSV) vectors. Curr. Opin. Virol. 2016, 21, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Skepper, J.N.; Whiteley, A.; Browne, H.; Minson, A. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment ⟶ deenvelopment ⟶ reenvelopment pathway. J. Virol. 2001, 75, 5697–5702. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Smith, G. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A. Assembly and Egress of an Alphaherpesvirus Clockwork. Adv. Anat. Embryol. Cell Biol. 2017, 223, 171–193. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Enquist, L.W. Directional spread of alphaherpesviruses in the nervous system. Viruses 2013, 5, 678–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diefenbach, R.J. Conserved tegument protein complexes: Essential components in the assembly of herpesviruses. Virus Res. 2015, 210, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.J.; Fraefel, C.; Cunningham, A.L.; Diefenbach, R.J. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 2009, 145, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Luxton, G.W.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5832–5837. [Google Scholar] [CrossRef] [PubMed]
- Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. Entry of pseudorabies virus: An immunogold-labeling study. J. Virol. 2005, 79, 3200–3205. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Smith, G.A. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J. Virol. 2010, 84, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Sandbaumhuter, M.; Dohner, K.; Schipke, J.; Binz, A.; Pohlmann, A.; Sodeik, B.; Bauerfeind, R. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 2013, 15, 248–269. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Buch, A.; Dohner, K.; Pohlmann, A.; Binz, A.; Prank, U.; Sandbaumhuter, M.; Bauerfeind, R.; Sodeik, B. Conserved Tryptophan Motifs in the Large Tegument Protein pUL36 Are Required for Efficient Secondary Envelopment of Herpes Simplex Virus Capsids. J. Virol. 2016, 90, 5368–5383. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and transport of herpes simplex virus type 1 in neurons: role of the cytoskeleton. Viruses 2018. submitted for publication. [Google Scholar]
- Khaitlina, S.Y. Intracellular transport based on actin polymerization. Biochemistry 2014, 79, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Woodrum, D.T.; Rich, S.A.; Pollard, T.D. Evidence for biased bidirectional polymerization of actin-filaments using heavy-meromyosin prepared by an improved method. J. Cell Biol. 1975, 67, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Svitkina, T. Filopodia initiation: Focus on the Arp2/3 complex and formins. Cell Adh. Migr. 2011, 5, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M.; Atkinson, S.J.; Ampe, C.; Vandekerckhove, J.; Pollard, T.D. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin-agarose. J. Cell Biol. 1994, 127, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.D.; Heuser, J.A.; Pollard, T.D. The interaction of Arp2/3 complex with actin: Nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA 1998, 95, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Pollard, T.D.; Beltzner, C.C. Structure and function of the Arp2/3 complex. Curr. Opin. Struct. Biol. 2002, 12, 768–774. [Google Scholar] [CrossRef]
- Volkmann, N.; Amann, K.J.; Stoilova-McPhie, S.; Egile, C.; Winter, D.C.; Hazelwood, L.; Heuser, J.E.; Li, R.; Pollard, T.D.; Hanein, D. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science 2001, 293, 2456–2459. [Google Scholar] [CrossRef] [PubMed]
- Hetrick, B.; Han, M.S.; Helgeson, L.A.; Nolen, B.J. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem. Biol. 2013, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Rouiller, I.; Xu, X.P.; Amann, K.J.; Egile, C.; Nickell, S.; Nicastro, D.; Li, R.; Pollard, T.D.; Volkmann, N.; Hanein, D. The structural basis of actin filament branching by the Arp2/3 complex. J. Cell Biol. 2008, 180, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Pollitt, A.Y.; Insall, R.H. WASP and SCAR/WAVE proteins: The drivers of actin assembly. J. Cell Sci. 2009, 122, 2575–2578. [Google Scholar] [CrossRef] [PubMed]
- Goley, E.D.; Rodenbusch, S.E.; Martin, A.C.; Welch, M.D. Critical conformational changes in the Arp2/3 complex are induced by nucleotide and nucleation promoting factor. Mol. Cell 2004, 16, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Miki, H.; Takenawa, T. Direct binding of the verprolin-homology domain in N-WASP to actin is essential for cytoskeletal reorganization. Biochem. Biophys. Res. Commun. 1998, 243, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 2011, 9, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.D.; Rosenblatt, J.; Skoble, J.; Portnoy, D.A.; Mitchison, T.J. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science 1998, 281, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Frischknecht, F.; Cudmore, S.; Moreau, V.; Reckmann, I.; Rottger, S.; Way, M. Tyrosine phosphorylation is required for actin-based motility of vaccinia but not Listeria or Shigella. Curr. Biol. 1999, 9, 89–92. [Google Scholar] [CrossRef]
- Mingo, R.M.; Han, J.; Newcomb, W.W.; Brown, J.C. Replication of herpes simplex virus: Egress of progeny virus at specialized cell membrane sites. J. Virol. 2012, 86, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Tiwari, V.; Shukla, D. Herpes simplex virus type 1 induces filopodia in differentiated P19 neural cells to facilitate viral spread. Neurosci. Lett. 2008, 440, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; Guay, G.; Lippé, R. Comprehensive Characterization of Extracellular Herpes Simplex Virus Type 1 Virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef] [PubMed]
- Feierbach, B.; Piccinotti, S.; Bisher, M.; Denk, W.; Enquist, L.W. Alpha-herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog. 2006, 2, e85. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Hutchinson, J.A.; Jackson, W.; Grose, C. Egress of light particles among filopodia on the surface of Varicella-Zoster virus-infected cells. J. Virol. 2008, 82, 2821–2835. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Desloges, N.; Rahaus, M.; Wolff, M.H. Varicella-zoster virus infection influences expression and organization of actin and alpha-tubulin but does not affect lamin A and vimentin. Intervirology 2005, 48, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Drebert, Z.; Golke, A.; Cymerys, J.; Slonska, A.; Chmielewska, A.; Tucholska, A.; Banbura, M.W. Equid herpesvirus type 1 (EHV-1) disrupts actin cytoskeleton during productive infection in equine leukocytes. Pol. J. Vet. Sci. 2015, 18, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slonska, A.; Cymerys, J.; Godlewski, M.M.; Dzieciatkowski, T.; Tucholska, A.; Chmielewska, A.; Golke, A.; Banbura, M.W. Equine herpesvirus type 1 (EHV-1)-induced rearrangements of actin filaments in productively infected primary murine neurons. Arch. Virol. 2014, 159, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Baines, J.D. Actin in herpesvirus infection. Viruses 2011, 3, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.J.; Akhtar, J.; Desai, P.; Shukla, D. A role for heparan sulfate in viral surfing. Biochem. Biophys. Res. Commun. 2010, 391, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Mues, M.B.; Cheshenko, N.; Wilson, D.W.; Gunther-Cummins, L.; Herold, B.C. Dynasore disrupts trafficking of herpes simplex virus proteins. J. Virol. 2015, 89, 6673–6684. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Xiang, Y.F.; Chen, J.N.; Lu, C.H.; Hao, J.; Du, Q.; Lai, C.C.; Qu, C.; Li, S.; Ju, H.Q.; et al. Pentagalloylglucose downregulates cofilin1 and inhibits HSV-1 infection. Antivir. Res. 2011, 89, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zheng, K.; Ju, H.; Wang, S.; Pei, Y.; Ding, W.; Chen, Z.; Wang, Q.; Qiu, X.; Zhong, M.; et al. Cofilin 1-mediated biphasic F-actin dynamics of neuronal cells affect herpes simplex virus 1 infection and replication. J. Virol. 2012, 86, 8440–8451. [Google Scholar] [CrossRef] [PubMed]
- Petermann, P.; Haase, I.; Knebel-Morsdorf, D. Impact of Rac1 and Cdc42 signaling during early herpes simplex virus type 1 infection of keratinocytes. J. Virol. 2009, 83, 9759–9772. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, S.; Schelhaas, M.; Jaeger, V.; Liebig, T.; Petermann, P.; Knebel-Morsdorf, D. Early herpes simplex virus type 1 infection is dependent on regulated Rac1/Cdc42 signalling in epithelial MDCKII cells. J. Gen. Virol. 2006, 87, 3483–3494. [Google Scholar] [CrossRef] [PubMed]
- Sakisaka, T.; Ikeda, W.; Ogita, H.; Fujita, N.; Takai, Y. The roles of nectins in cell adhesions: Cooperation with other cell adhesion molecules and growth factor receptors. Curr. Opin. Cell Biol. 2007, 19, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.; Tiwari, V.; Scanlan, P.M.; Valyi-Nagy, T.; Yue, B.Y.; Shukla, D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 2006, 174, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Burnham, L.; Thompson, J.M.; Shukla, D.; Tiwari, V. Role of Filopodia in HSV-1 Entry into Zebrafish 3-O-Sulfotransferase-3-Expressing Cells. Open Virol. J. 2013, 7, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Devadas, D.; Koithan, T.; Diestel, R.; Prank, U.; Sodeik, B.; Dohner, K. Herpes simplex virus internalization into epithelial cells requires Na+/H+ exchangers and p21-activated kinases but neither clathrin- nor caveolin-mediated endocytosis. J. Virol. 2014, 88, 13378–13395. [Google Scholar] [CrossRef] [PubMed]
- Favoreel, H.W.; Van Minnebruggen, G.; Adriaensen, D.; Nauwynck, H.J. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. USA 2005, 102, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeke, C.; Deruelle, M.; Nauwynck, H.J.; Coller, K.E.; Smith, G.A.; Van Doorsselaere, J.; Favoreel, H.W. The kinase activity of pseudorabies virus US3 is required for modulation of the actin cytoskeleton. Virology 2009, 385, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeke, C.; Radu, M.; Deruelle, M.; Nauwynck, H.; Hofmann, C.; Jaffer, Z.M.; Chernoff, J.; Favoreel, H.W. Alphaherpesvirus US3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc. Natl. Acad. Sci. USA 2009, 106, 8707–8712. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Roy, B.B.; Zhang, H.; Banfield, B.W. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 2010, 397, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.; Van den Broeke, C.; Grauwet, K.; Baert, K.; Claessen, C.; De Pelsmaeker, S.; Van Waesberghe, C.; Favoreel, H.W. Pseudorabies virus US3 leads to filamentous actin disassembly and contributes to viral genome delivery to the nucleus. Vet. Microbiol. 2015, 177, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.; Van den Broeke, C.; Van Waesberghe, C.; Van Troys, L.; Favoreel, H.W. Pseudorabies virus US3 triggers RhoA phosphorylation to reorganize the actin cytoskeleton. J. Gen. Virol. 2015, 96, 2328–2335. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y. Us3, a multifunctional protein kinase encoded by herpes simplex virus 1: How does it function in vivo? Cornea 2013, 32 (Suppl. S1), S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Banfield, B.W. Subcellular localization of the alphaherpesvirus serine/threonine kinase Us3 as a determinant of Us3 function. Virulence 2010, 1, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Tischer, B.K.; Trapp, S.; Osterrieder, N. The protein encoded by the US3 orthologue of Marek's disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J. Virol. 2005, 79, 3987–3997. [Google Scholar] [CrossRef] [PubMed]
- Ladelfa, M.F.; Kotsias, F.; Del Medico Zajac, M.P.; Van den Broeke, C.; Favoreel, H.; Romera, S.A.; Calamante, G. Effect of the US3 protein of bovine herpesvirus 5 on the actin cytoskeleton and apoptosis. Vet. Microbiol. 2011, 153, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Proft, A.; Spiesschaert, B.; Izume, S.; Taferner, S.; Lehmann, M.J.; Azab, W. The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeke, C.; Radu, M.; Nauwynck, H.J.; Chernoff, J.; Favoreel, H.W. Role of group A p21-activated kinases in the anti-apoptotic activity of the pseudorabies virus US3 protein kinase. Virus Res. 2011, 155, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Lamote, J.A.; Glorieux, S.; Nauwynck, H.J.; Favoreel, H.W. The US3 Protein of Pseudorabies Virus Drives Viral Passage across the Basement Membrane in Porcine Respiratory Mucosa Explants. J. Virol. 2016, 90, 10945–10950. [Google Scholar] [CrossRef] [PubMed]
- Richerioux, N.; Blondeau, C.; Wiedemann, A.; Remy, S.; Vautherot, J.F.; Denesvre, C. Rho-ROCK and Rac-PAK signaling pathways have opposing effects on the cell-to-cell spread of Marek’s Disease Virus. PLoS ONE 2012, 7, e44072. [Google Scholar] [CrossRef] [PubMed]
- Masters, T.A.; Kendrick-Jones, J.; Buss, F. Myosins: Domain Organisation, Motor Properties, Physiological Roles and Cellular Functions. Handb. Exp. Pharmacol. 2017, 235, 77–122. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.A.; Spudich, J.A. The myosin superfamily at a glance. J. Cell Sci. 2012, 125, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.F.; Langford, G.M. Myosin superfamily evolutionary history. Anat. Rec. 2002, 268, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Baines, J.D. Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J. Virol. 2010, 84, 9889–9896. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, H.; Elliott, G.; O’Hare, P. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J. Virol. 2002, 76, 3471–3481. [Google Scholar] [CrossRef] [PubMed]
- Antoine, T.E.; Shukla, D. Inhibition of myosin light chain kinase can be targeted for the development of new therapies against herpes simplex virus type-1 infection. Antivir. Ther. 2014, 19, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Arii, J.; Goto, H.; Suenaga, T.; Oyama, M.; Kozuka-Hata, H.; Imai, T.; Minowa, A.; Akashi, H.; Arase, H.; Kawaoka, Y.; et al. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 2010, 467, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Arii, J.; Hirohata, Y.; Kato, A.; Kawaguchi, Y. Nonmuscle myosin heavy chain IIb mediates herpes simplex virus 1 entry. J. Virol. 2015, 89, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Gao, S.J. Actin dynamics regulate multiple endosomal steps during Kaposi’s sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog. 2009, 5, e1000512. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.J.; Prod’homme, V.; Purbhoo, M.A.; Moore, M.; Aicheler, R.J.; Heinzmann, M.; Bailer, S.M.; Haas, J.; Antrobus, R.; Weekes, M.P.; et al. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 2014, 16, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, A.R.; Lawler, J.L.; Coen, D.M. A role for nuclear F-actin induction in human cytomegalovirus nuclear egress. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Hogue, I.B.; Feric, M.; Thiberge, S.Y.; Sodeik, B.; Brangwynne, C.P.; Enquist, L.W. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc. Natl. Acad. Sci. USA 2015, 112, E5725–E5733. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Virding, S.; Thiberge, S.Y.; Scherer, J.; Wodrich, H.; Ruzsics, Z.; Koszinowski, U.H.; Enquist, L.W. Nuclear herpesvirus capsid motility is not dependent on F-actin. MBio 2014, 5, e01909–e01914. [Google Scholar] [CrossRef] [PubMed]
- Jean Beltran, P.M.; Mathias, R.A.; Cristea, I.M. A Portrait of the Human Organelle Proteome In Space and Time during Cytomegalovirus Infection. Cell Syst. 2016, 3, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Cymerys, J.; Slonska, A.; Skwarska, J.; Banbura, M.W. Function of myosin during entry and egress of equid herpesvirus type 1 in primary murine neurons. Acta Virol. 2016, 60, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.B.; Turner, R.; Stevenson, P.G.; Way, M. KSHV-TK is a tyrosine kinase that disrupts focal adhesions and induces Rho-mediated cell contraction. EMBO J. 2015, 34, 448–465. [Google Scholar] [CrossRef] [PubMed]
- Michael, K.; Klupp, B.G.; Mettenleiter, T.C.; Karger, A. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J. Virol. 2006, 80, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Kollman, J.M.; Polka, J.K.; Zelter, A.; Davis, T.N.; Agard, D.A. Microtubule nucleating gamma-TuSC assembles structures with 13-fold microtubule-like symmetry. Nature 2010, 466, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.A.; Shalini, S.; Risk, J.M.; Day, C.L.; Kumar, S. A direct interaction with NEDD1 regulates gamma-tubulin recruitment to the centrosome. PLoS ONE 2010, 5, e9618. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Portilho, D.M.; Persson, R.; Arhel, N. Role of non-motile microtubule-associated proteins in virus trafficking. Biomol. Concepts 2016, 7, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Pasdeloup, D.; Labetoulle, M.; Rixon, F.J. Differing effects of herpes simplex virus 1 and pseudorabies virus infections on centrosomal function. J. Virol. 2013, 87, 7102–7112. [Google Scholar] [CrossRef] [PubMed]
- Kotsakis, A.; Pomeranz, L.E.; Blouin, A.; Blaho, J.A. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 2001, 75, 8697–8711. [Google Scholar] [CrossRef] [PubMed]
- Dienes, H.P.; Hiller, G.; Muller, S.; Falke, D. Microtubules and intermediate filaments of herpes simplex virus infected cells. Arch. Virol. 1987, 94, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, A.; Rychlowski, M.; Lipinska, A.D.; Bienkowska-Szewczyk, K. Point mutations in BHV-1 Us3 gene abolish its ability to induce cytoskeletal changes in various cell types. Vet. Microbiol. 2010, 143, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Jovasevic, V.; Naghavi, M.H.; Walsh, D. Microtubule plus end-associated CLIP-170 initiates HSV-1 retrograde transport in primary human cells. J. Cell Biol. 2015, 211, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.H.; Gundersen, G.G.; Walsh, D. Plus-end tracking proteins, CLASPs, and a viral Akt mimic regulate herpesvirus-induced stable microtubule formation and virus spread. Proc. Natl. Acad. Sci. USA 2013, 110, 18268–18273. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Schmidt, E.E.; Halford, W.P. ICP0 dismantles microtubule networks in herpes simplex virus-infected cells. PLoS ONE 2010, 5, e10975. [Google Scholar] [CrossRef] [PubMed]
- Cheishvili, D.; Maayan, C.; Cohen-Kupiec, R.; Lefler, S.; Weil, M.; Ast, G.; Razin, A. IKAP/Elp1 involvement in cytoskeleton regulation and implication for familial dysautonomia. Hum. Mol. Genet. 2011, 20, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.J.; Diefenbach, E.; Fraefel, C.; Diefenbach, R.J. Identification of host cell proteins which interact with herpes simplex virus type 1 tegument protein pUL37. Biochem. Biophys. Res. Commun. 2012, 417, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Marozin, S.; Prank, U.; Sodeik, B. Herpes simplex virus type 1 infection of polarized epithelial cells requires microtubules and access to receptors present at cell-cell contact sites. J. Gen. Virol. 2004, 85, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Mabit, H.; Nakano, M.Y.; Prank, U.; Saam, B.; Dohner, K.; Sodeik, B.; Greber, U.F. Intact microtubules support adenovirus and herpes simplex virus infections. J. Virol. 2002, 76, 9962–9971. [Google Scholar] [CrossRef] [PubMed]
- Topp, K.S.; Meade, L.B.; LaVail, J.H. Microtubule polarity in the peripheral processes of trigeminal ganglion cells: Relevance for the retrograde transport of herpes simplex virus. J. Neurosci. 1994, 14, 318–325. [Google Scholar] [PubMed]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Tanaka, Y. Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Exp. Cell Res. 2015, 334, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Hook, P.; Vallee, R.B. The dynein family at a glance. J. Cell Sci. 2006, 119, 4369–4371. [Google Scholar] [CrossRef] [PubMed]
- Allan, V.J. Cytoplasmic dynein. Biochem. Soc. Trans. 2011, 39, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Enquist, L.W. Axonal spread of neuroinvasive viral infections. Trends Microbiol. 2015, 23, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Dohner, K.; Nagel, C.H.; Sodeik, B. Viral stop-and-go along microtubules: Taking a ride with dynein and kinesins. Trends Microbiol. 2005, 13, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P.; Way, M. Coupling viruses to dynein and kinesin-1. EMBO J. 2011, 30, 3527–3539. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Gross, S.P.; Enquist, L.W. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 3466–3470. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, C.J.; Dawe, R.K.; Christie, K.R.; Cleveland, D.W.; Dawson, S.C.; Endow, S.A.; Goldstein, L.S.; Goodson, H.V.; Hirokawa, N.; Howard, J.; et al. A standardized kinesin nomenclature. J. Cell Biol. 2004, 167, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.J.; Kon, T.; Knight, P.J.; Sutoh, K.; Burgess, S.A. Functions and mechanics of dynein motor proteins. Nat. Rev. Mol. Cell Biol. 2013, 14, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Bhabha, G.; Johnson, G.T.; Schroeder, C.M.; Vale, R.D. How Dynein Moves Along Microtubules. Trends Biochem. Sci. 2016, 41, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Vaughan, K.T.; Vallee, R.B.; Roizman, B. The herpes simplex virus 1 U(L)34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 2000, 74, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Miranda-Saksena, M.; Diefenbach, E.; Holland, D.J.; Boadle, R.A.; Armati, P.J.; Cunningham, A.L. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 2002, 76, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Shanda, S.K.; Wilson, D.W. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 2008, 82, 7388–7394. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef] [PubMed]
- Zaichick, S.V.; Bohannon, K.P.; Hughes, A.; Sollars, P.J.; Pickard, G.E.; Smith, G.A. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 2013, 13, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Pasdeloup, D.; McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J. Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J. Virol. 2013, 87, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J.; Pasdeloup, D. Dystonin/BPAG1 promotes plus-end-directed transport of herpes simplex virus 1 capsids on microtubules during entry. J. Virol. 2013, 87, 11008–11018. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Davis, A.; Miranda-Saksena, M.; Fernandez, M.A.; Kelly, B.J.; Jones, C.A.; LaVail, J.H.; Xue, J.; Lai, J.; Cunningham, A.L. The Basic Domain of Herpes Simplex Virus 1 pUS9 Recruits Kinesin-1 To Facilitate Egress from Neurons. J. Virol. 2016, 90, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Daniel, G.R.; Sollars, P.J.; Pickard, G.E.; Smith, G.A. Pseudorabies Virus Fast Axonal Transport Occurs by a pUS9-Independent Mechanism. J. Virol. 2015, 89, 8088–8091. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Greco, T.M.; Taylor, M.P.; Ambrosini, A.E.; Cristea, I.M.; Enquist, L.W. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 2012, 12, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Dohner, K.; Radtke, K.; Schmidt, S.; Sodeik, B. Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26. J. Virol. 2006, 80, 8211–8224. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Shubeita, G.T.; Coller, K.E.; Lee, J.I.; Haverlock-Moyns, S.; Gross, S.P.; Smith, G.A. The Herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J. Virol. 2006, 80, 5494–5498. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.W.; Diefenbach, R.J.; Homa, F.L.; Miranda-Saksena, M.; Rixon, F.J.; Vittone, V.; Byth, K.; Cunningham, A.L. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 2004, 279, 28522–28530. [Google Scholar] [CrossRef] [PubMed]
- Daniel, G.R.; Sollars, P.J.; Pickard, G.E.; Smith, G.A. The pseudorabies virus protein, pUL56, enhances virus dissemination and virulence but is dispensable for axonal transport. Virology 2016, 488, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Kawaguchi, Y.; Nishiyama, Y. Herpes simplex virus type 2 membrane protein UL56 associates with the kinesin motor protein KIF1A. J. Gen. Virol. 2005, 86, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Luxton, G.W.; Lee, J.I.; Haverlock-Moyns, S.; Schober, J.M.; Smith, G.A. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 2006, 80, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Wolfstein, A.; Nagel, C.H.; Radtke, K.; Dohner, K.; Allan, V.J.; Sodeik, B. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 2006, 7, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Sollars, P.J.; Pitts, J.D.; Stults, A.M.; Heldwein, E.E.; Pickard, G.E.; Smith, G.E. The pUL37 tegument protein guides alpha-herpesvirus retrograde axonal transport to promote neuroinvasion. PLoS Pathog. 2017. [Google Scholar] [CrossRef] [PubMed]
- Buch, A.; Muller, O.; Ivanova, L.; Dohner, K.; Bialy, D.; Bosse, J.B.; Pohlmann, A.; Binz, A.; Hegemann, M.; Nagel, C.H.; et al. Inner tegument proteins of Herpes Simplex Virus are sufficient for intracellular capsid motility in neurons but not for axonal targeting. PLoS Pathog. 2017, 13, e1006813. [Google Scholar] [CrossRef] [PubMed]
- Pernigo, S.; Lamprecht, A.; Steiner, R.A.; Dodding, M.P. Structural basis for kinesin-1:cargo recognition. Science 2013, 340, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P.; Mitter, R.; Humphries, A.C.; Way, M. A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 2011, 30, 4523–4538. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.W.; Lin, S.J.; Tsai, S.C.; Lin, J.H.; Chen, M.R.; Wang, J.T.; Lee, C.P.; Tsai, C.H. Regulation of microtubule dynamics through phosphorylation on stathmin by Epstein-Barr virus kinase BGLF4. J. Biol. Chem. 2010, 285, 10053–10063. [Google Scholar] [CrossRef] [PubMed]
- Curmi, P.A.; Gavet, O.; Charbaut, E.; Ozon, S.; Lachkar-Colmerauer, S.; Manceau, V.; Siavoshian, S.; Maucuer, A.; Sobel, A. Stathmin and its phosphoprotein family: General properties, biochemical and functional interaction with tubulin. Cell Struct. Funct. 1999, 24, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Smith, M.S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J. Virol. 2005, 79, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Kratchmarov, R.; Kramer, T.; Greco, T.M.; Taylor, M.P.; Ch’ng, T.H.; Cristea, I.M.; Enquist, L.W. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J. Virol. 2013, 87, 9431–9440. [Google Scholar] [CrossRef] [PubMed]
- Howard, P.W.; Howard, T.L.; Johnson, D.C. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J. Virol. 2013, 87, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.; Kuczmarski, E.R.; Herrmann, H.; Goldman, R.D. Intermediate Filaments Play a Pivotal Role in Regulating Cell Architecture and Function. J. Biol. Chem. 2015, 290, 17145–17153. [Google Scholar] [CrossRef] [PubMed]
- Godsel, L.M.; Hobbs, R.P.; Green, K.J. Intermediate filament assembly: Dynamics to disease. Trends Cell Biol. 2008, 18, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Cleland, M.M.; Murthy, S.N.; Mahammad, S.; Kuczmarski, E.R. Inroads into the structure and function of intermediate filament networks. J. Struct. Biol. 2012, 177, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Aebi, U. Intermediate filaments: A dynamic network that controls cell mechanics. F1000Prime Rep. 2014, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Ku, N.O.; Tao, G.Z.; Toivola, D.M.; Liao, J. “Heads and tails” of intermediate filament phosphorylation: Multiple sites and functional insights. Trends Biochem. Sci. 2006, 31, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Hertel, L. Onset of human cytomegalovirus replication in fibroblasts requires the presence of an intact vimentin cytoskeleton. J. Virol. 2009, 83, 7015–7028. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Goshima, F.; Nishizawa, Y.; Daikoku, T.; Takakuwa, H.; Ohtsuka, K.; Yoshikawa, T.; Nishiyama, Y. Phosphorylation of cytokeratin 17 by herpes simplex virus type 2 US3 protein kinase. Microbiol. Immunol. 2002, 46, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Norregard Nielsen, L.; Forchhammer, J.; Dabelsteen, E.; Jepsen, A.; Stubbe Teglbjaerg, C.; Norrild, B. Herpes simplex virus-induced changes of the keratin type intermediate filament in rat epithelial cells. J. Gen. Virol. 1987, 68, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.E.; Liang, L.; Baines, J.D. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J. Virol. 2004, 78, 5564–5575. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Haas, J.; Wagner, M.; Krohne, G.; Koszinowski, U.H. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 2002, 297, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Leigh, K.E.; Sharma, M.; Mansueto, M.S.; Boeszoermenyi, A.; Filman, D.J.; Hogle, J.M.; Wagner, G.; Coen, D.M.; Arthanari, H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9010–9015. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kamil, J.P.; Coughlin, M.; Reim, N.I.; Coen, D.M. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J. Virol. 2014, 88, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Muller, F.; Granzow, H.; Klupp, B.G. The way out: What we know and do not know about herpesvirus nuclear egress. Cell. Microbiol. 2013, 15, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kato, A.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kawaguchi, Y. Role of Host Cell p32 in Herpes Simplex Virus 1 De-Envelopment during Viral Nuclear Egress. J. Virol. 2015, 89, 8982–8998. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the γ134.5 Protein That Facilitates Nuclear Egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef] [PubMed]
- Mou, F.; Forest, T.; Baines, J.D. US3 of Herpes Simplex Virus Type 1 Encodes a Promiscuous Protein Kinase That Phosphorylates and Alters Localization of Lamin A/C in Infected Cells. J. Virol. 2007, 81, 6459–6470. [Google Scholar] [CrossRef] [PubMed]
- Cano-Monreal, G.L.; Wylie, K.M.; Cao, F.; Tavis, J.E.; Morrison, L.A. Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology 2009, 392, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.B.; Hofemeister, H.; O’Hare, P. Herpes simplex virus infection induces phosphorylation and delocalization of emerin, a key inner nuclear membrane protein. J. Virol. 2007, 81, 4429–4437. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.O.; Arvin, A.M. Microarray analysis of host cell gene transcription in response to varicella-zoster virus infection of human T cells and fibroblasts in vitro and SCIDhu skin xenografts in vivo. J. Virol. 2003, 77, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Esclatine, A.; Roizman, B. The patterns of accumulation of cellular RNAs in cells infected with a wild-type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. Proc. Natl. Acad. Sci. USA 2002, 99, 17031–17036. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.; Enquist, L.W. Transcriptional response of a common permissive cell type to infection by two diverse alphaherpesviruses. J. Virol. 2004, 78, 3489–3501. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Surma, M.; Shi, S.; Lambert-Cheatham, N.; Shi, J. Novel Insights into the Roles of Rho Kinase in Cancer. Arch. Immunol. Ther. Exp. 2016, 64, 259–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, G.; Tatrai, P.; Gergely, F. Hitting the brakes: Targeting microtubule motors in cancer. Br. J. Cancer 2015, 113, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Eira, J.; Silva, C.S.; Sousa, M.M.; Liz, M.A. The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog. Neurobiol. 2016, 141, 61–82. [Google Scholar] [CrossRef] [PubMed]
- Elion, G.B.; Furman, P.A.; Fyfe, J.A.; de Miranda, P.; Beauchamp, L.; Schaeffer, H.J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. USA 1977, 74, 5716–5720. [Google Scholar] [CrossRef] [PubMed]
- Hukkanen, V. Herpesvirus vectors in gene therapy. Open Virol. J. 2010, 4, 94–95. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Virus | Viral Protein | Host Cell Proteins/System | Role | Reference |
|---|---|---|---|---|
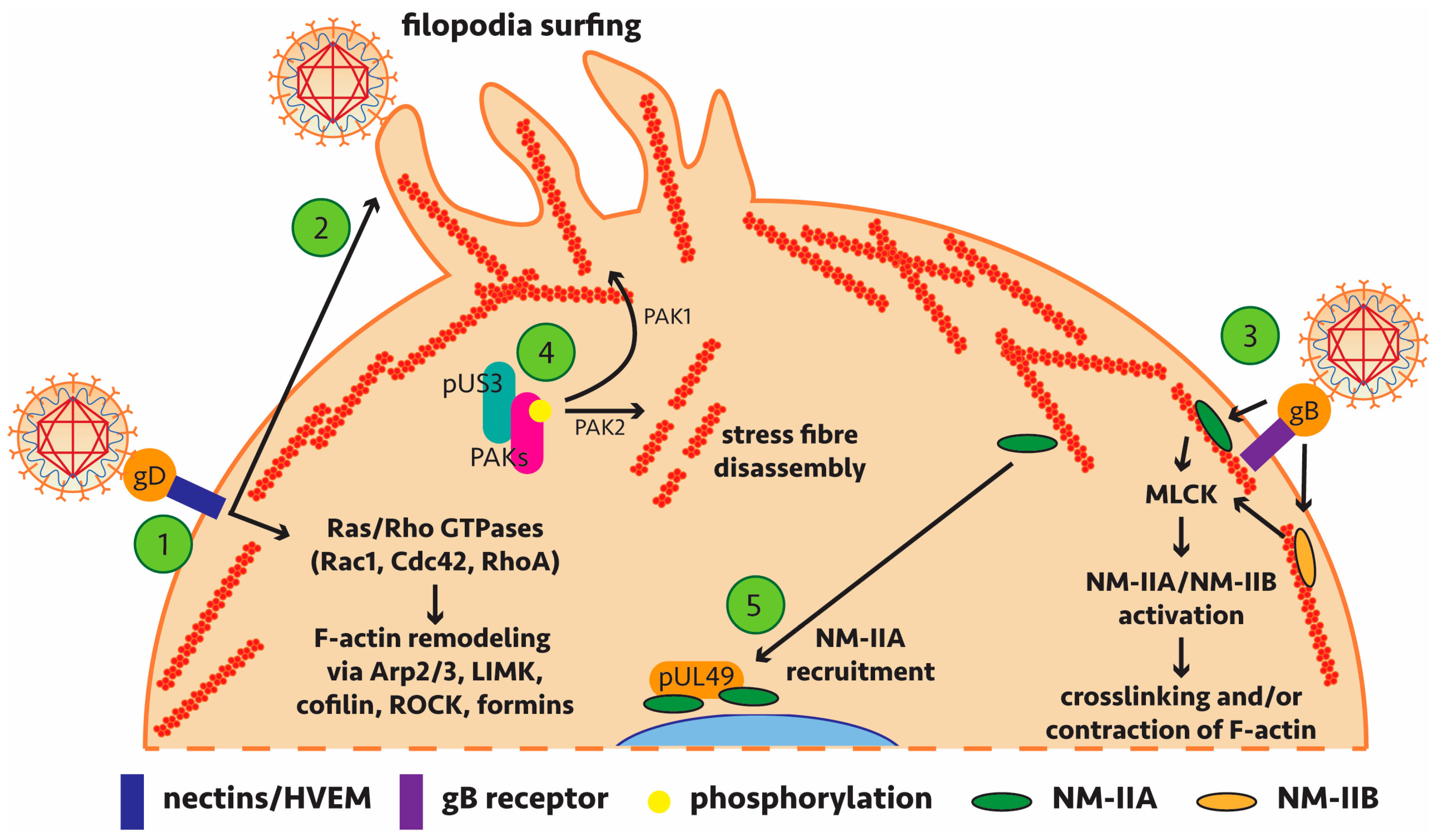
| HSV-1 HSV-2 | gD | Nectins | gD binds to host-cell receptor nectin. Nectins regulate actin reorganisation by activating remodelling proteins like Ras/Rho GTPases (Rap1, Cdc42, Rac1). Rac1/Cdc42 have been implicated in signalling during early HSV-1 infection. However, evidence has shown that Rac1 and Cdc42 signalling does not occur in infected keratinocytes [75]. | [76,77,78] |
| HSV-1 | gD | Likely nectin-1/HVEM | Following viral binding, there is activation of Cdc42 and RhoA, causing filopodium-like protrusions in corneal fibroblasts and nectin-1-expressing Chinese hamster ovary (CHO) cells. Virus associates with these protrusions during viral entry and actin depolymerisation drugs inhibit viral entry. Also observed in a zebrafish model. | [78,79] |
| HSV-1 | Unknown | Na+/H+ exchangers (NHE), p21-activated kinases | Internalisation of HSV-1 relies on the activity of these NHEs on the plasma membrane of Vero, HeLa, HEp-2 and PtK2 cells. These are known to be involved in macropinocytosis, an actin-dependent endocytic process which takes up extracellular fluid and macromolecules. This process can withstand the endocytosis of large structures (0.2–5 μm) which a large pathogen like HSV-1 can exploit. | [80] |
| HSV-2 PrV | pUS3 | PAK1/PAK2 | pUS3 directly phosphorylates group A p21-activated kinases (PAKs). Actin stress fibre disassembly during PrV infection of mouse embryonic fibroblast (MEF) and swine testicle (ST) cells is pUS3-mediated and requires PAK2. Cellular projections are mediated by PAK1. pUS3 kinase activity leads to protein kinase A-dependent phosphorylation of RhoA in ST cells; this subverts the antagonistic RhoA and Cdc42/Rac1/PAK signalling cascades for actin remodelling. | [81,82,83,84,85,86] |
| Virus | Viral Protein | Host-Cell Proteins/System | Role | Reference |
|---|---|---|---|---|
| HSV-1 | Unknown | Myosin Va | Myosin Va is activated during infection, facilitating transport of virion- and glycoprotein-bearing vesicles from TGN to plasma membrane through cortical actin in HeLa cells. It is hypothesised that egressing virions (collected within TGN-derived vesicles) behave in a similar manner to other myosin-dependent cargo: kinesin motors (see Section 4) deliver vesicles to cortical actin and myosin Va “captures” the vesicles and then transports them to the plasma membrane. | [98] |
| HSV-1 | pUL49 (VP22) | Non-muscle myosin heavy chain IIA (NM-IIA) | Affinity chromatography experiments with HSV-1-infected baby hamster kidney (BHK) cell extracts have shown tegument protein VP22 interacts with NM-IIA. HSV-1 infection of Vero cells redistributes NM-IIA but only a subpopulation of NM-IIA colocalises with VP22 in a perinuclear cluster. | [99] |
| HSV-1 | gB | NM-IIA/myosin light chain kinase (MLCK) | NM-IIA is a functional coreceptor for gB in Vero cells. Inhibition of NM-IIA (by blebbistatin) and MLCK (by ML-7 and ML-9) decreased viral entry into corneal epithelial cells. Activation of NM-IIA by MLCK is necessary for the cytoskeletal rearrangements needed for HSV-1 infection of corneal cells. To regulate actin, NM-IIA cross-links and contracts F-actin. | [100,101] |
| HSV-1 | gB | Non-muscle myosin heavy chain IIB (NM-IIB) | Interaction may serve as an entry coreceptor in the CV-1 in origin with SV40 genes (COS) cell line as above. Activation of NM-IIB by MLCK is also necessary for the cytoskeletal rearrangements. Likely to be important in a range of cell types. | [102] |
| Virus | Viral Protein | Host-Cell Proteins/System | Role | Reference |
|---|---|---|---|---|
| HSV-1 | Capsid (unknown) | Microtubule plus–end tracking protein (+TIP) complex EB1, CLIP-170 and dynactin-1 | Studies in normal human dermal fibroblasts (NHDFs) show EB1 directs viral capsid interaction with plus end of microtubules. Stabilises microtubules and recruits molecular motor dynein for retrograde transport during initial viral entry. | [121] |
| HSV-1 | pUS3 | Glycogen synthase kinase 3β | pUS3 phosphorylation inactivates the host cell kinase in NHDFs, leading to microtubule stabilisation by +TIP and cytoplasmic linker-associated proteins (CLASPs), to enhance viral spread. | [122] |
| HSV-1 | ICP0 | Unknown | ICP0 is a viral E3 ligase which was found to destabilise and unbundle microtubules in Vero cells to aid in viral assembly and egress. | [123] |
| HSV-1 | pUL37 | IKAP (Iκβ kinase complex associated protein) | Yeast two-hybrid screening indicated an interaction between tegument protein pUL37 and IKAP. IKAP has proposed roles in microtubule stabilisation [124]. pUL37 binding of the C-terminal region of IKAP could regulate its activity and stabilise cytoskeletal rearrangements during the changes that occur from infection to enhance viral replication. Yet to be tested in cell lines. | [125] |
| Virus | Viral Protein | Host-Cell Proteins/System | Role | Reference |
|---|---|---|---|---|
| HSV-1 | pUL34 | Intermediate chain of the dynein complex (IC-1a) | Pulldown experiments of infected Vero and HEp-2 cells with IC-1a (and reciprocal experiments) identified an interaction with pUL34. pUL34 localised to the nuclear membrane when expressed by a baculovirus vector, confirming the protein is involved in transport to the nuclear membrane in the viral context. pUL34 is not a structural protein [64] so unlikely to be involved in cytoplasmic viral capsid transport. | [140] |
| HSV-1 | pUS11 | Kinesin-1 (KIF5) | Residues 867–894 of ubiquitous human kinesin-1 bind to a C-terminal RNA-binding domain of tegument pUS11 as evidenced by pulldown assays. HSV-1 pUS11 has 63% homology to HSV-2 pUS11, with variation in the N-terminal half, so this interaction could prove to be transferable. Not confirmed in vivo and one study suggests pUS11 is not a structural tegument protein [64]. | [141] |
| HSV-1 | Tegument proteins | Dynein, dynactin, kinesin-1 | Tegumented capsids (lacking outer tegument and envelopes) were capable of binding microtubule associated proteins (MAPs) sourced from pig brain cytosol. 10% of capsids tested by in vitro single particle analysis had bound dynein and kinesin-1 simultaneously, suggesting HSV-1 capsid transport is not directed by exclusive presence of either minus- or plus-ended motors. Inner tegument, pUL36 and pUL37, suggested as most likely to bind motors or recruit other tegument proteins that bind motors at this stage, especially with early findings that without pUL36, HSV-1 particles form but have reduced infectivity and a decreased ability to bind to and transport along microtubules [142]. | [143] |
| PrV | pUL36 | Dynein, dynactin | Immunoprecipitation of pUL36-transfected HEK293 cells showed that it interacts with dynein/dynactin and can drive transport in the absence of other viral proteins when transfected into Vero cells. pUL36 is capable of transporting viral capsid along microtubules in conjunction with capsid-binding pUL25. A large proline-rich domain in the pUL36 C-terminus contributes to the interaction. | [144] |
| HSV-1 | pUL37 | Dystonin/BPAG1 | Tegument protein pUL37 recruits dystonin/BPAG1 in human foetal foreskin fibroblasts (HFFF2), which most likely functions to crosslink and stabilise microtubules, to facilitate viral capsid transport during viral entry. Plus-end directed transport is inhibited by dystonin depletion, providing evidence that pUL37-dystonin interaction is required for transport of capsids from the centrosome to the nucleus. | [145,146] |
| HSV-1 | pUS9 | Kinesin-1 | Five arginine residues in the basic domain of envelope protein pUS9 bind host motor kinesin-1 as determined by truncation construct pulldown studies. This domain was shown to contribute to anterograde axonal transport in infected primary rat dorsal root ganglionic (DRG) neurons and a mouse zosteriform model. | [147] |
| PrV | pUS9 | Kinesin-3 (KIF1A) | pUS9 was found to interact with kinesin-3 using GFP-Trap pulldown. This interaction was shown to mediate efficient axonal sorting and anterograde axonal transport of viral particles in primary rat superior cervical ganglion neurons [148]. | [149] |
| HSV-1 | pUL35 (VP26) | Dynein light chains Tctex1 and RP3 | In vitro yeast two-hybrid evidence that capsid protein VP26 recruits these dynein light chains. Microinjection of HEp-2 cells with HSV-1 ± VP26 suggested VP26 was important for viral retrograde transport. Subsequent deletion studies in cell lines suggest this is a dispensable interaction [150,151]. | [152] |
| HSV-2 | pUL56 | Kinesin-3 (KIF1A) | In vitro evidence that envelope protein pUL56 interacts with kinesin-3 with a C-terminal transmembrane domain important for this interaction in transfected Vero cells. Possible role in anterograde axonal transport. Shown in PrV to support virus dissemination in vivo in embryonic chick DRG and an infected mouse model but is dispensable for intra-axonal transport beyond the sorting barrier [153]. | [154] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denes, C.E.; Miranda-Saksena, M.; Cunningham, A.L.; Diefenbach, R.J. Cytoskeletons in the Closet—Subversion in Alphaherpesvirus Infections. Viruses 2018, 10, 79. https://doi.org/10.3390/v10020079
Denes CE, Miranda-Saksena M, Cunningham AL, Diefenbach RJ. Cytoskeletons in the Closet—Subversion in Alphaherpesvirus Infections. Viruses. 2018; 10(2):79. https://doi.org/10.3390/v10020079
Chicago/Turabian StyleDenes, Christopher E., Monica Miranda-Saksena, Anthony L. Cunningham, and Russell J. Diefenbach. 2018. "Cytoskeletons in the Closet—Subversion in Alphaherpesvirus Infections" Viruses 10, no. 2: 79. https://doi.org/10.3390/v10020079
APA StyleDenes, C. E., Miranda-Saksena, M., Cunningham, A. L., & Diefenbach, R. J. (2018). Cytoskeletons in the Closet—Subversion in Alphaherpesvirus Infections. Viruses, 10(2), 79. https://doi.org/10.3390/v10020079