The Microbial Zoo in the C. elegans Intestine: Bacteria, Fungi and Viruses

Abstract
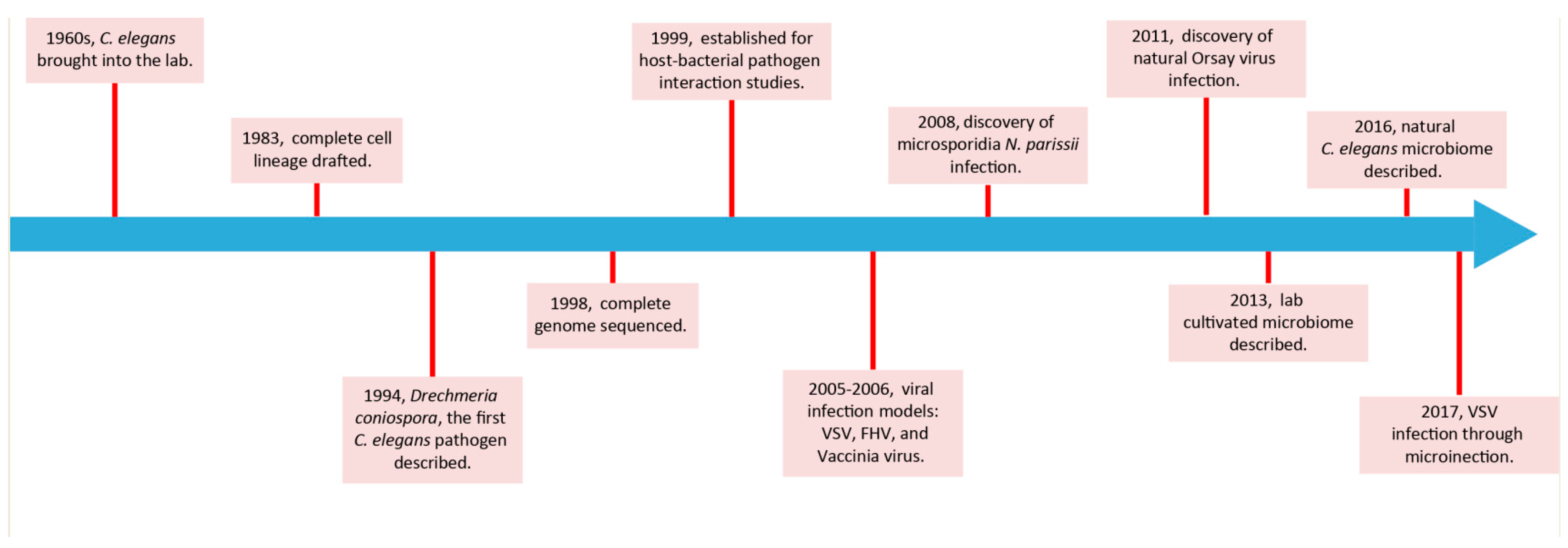
:1. Preface
2. The C. elegans Microbiome
3. Bacteria–Host Interactions in the C. elegans Gut
4. Fungi–Host Interactions in the C. elegans Gut
5. Virus–Host Interactions in the C. elegans Gut
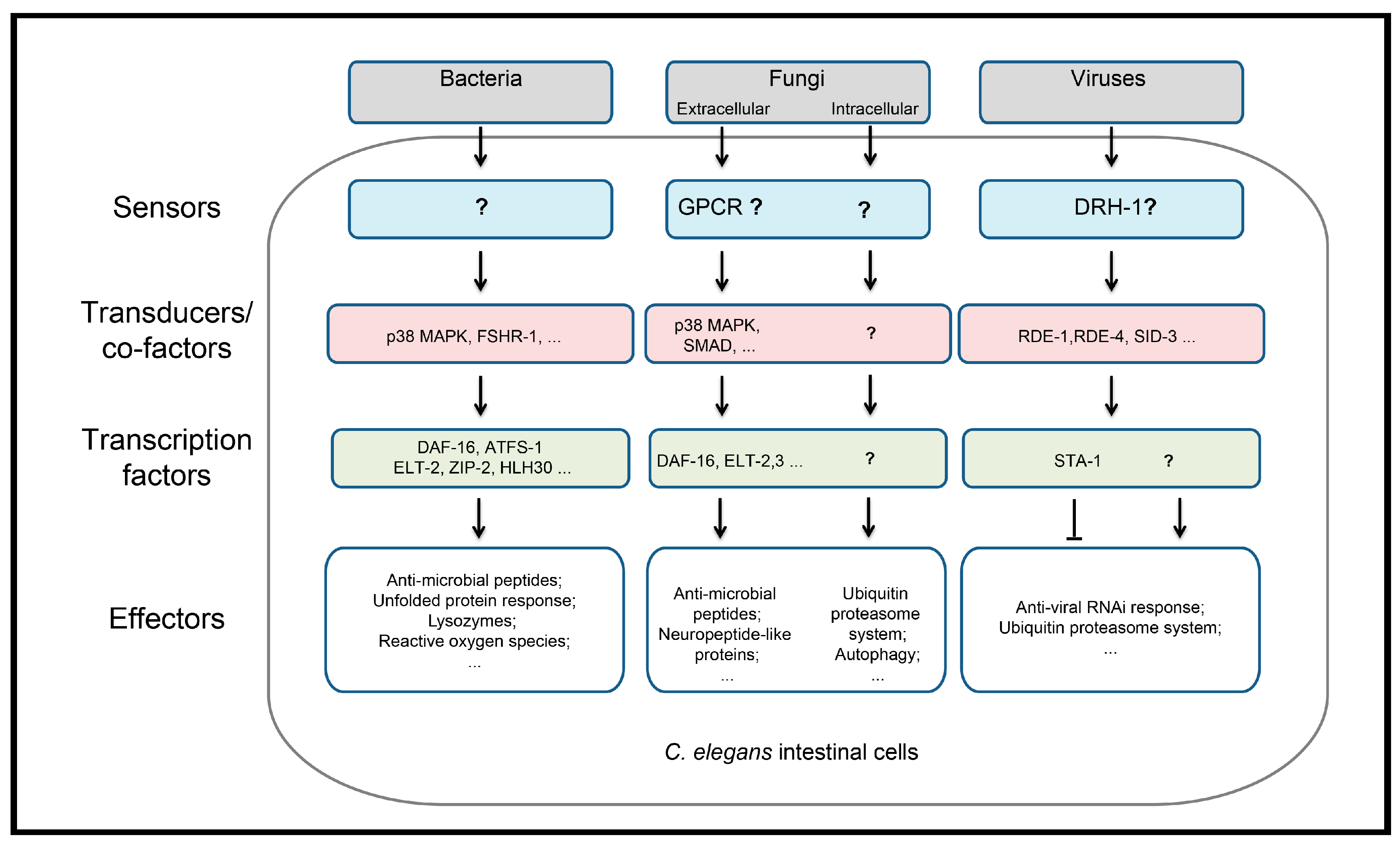
6. C. elegans as a Model to Study Microbial Trans-Kingdom Interactions
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Ellis, H.M.; Horvitz, H.R. Genetic control of programmed cell death in the nematode C. elegans. Cell 1986, 44, 817–829. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 1998, 282, 2012–2018. [Google Scholar]
- Troemel, E.R.; Felix, M.A.; Whiteman, N.K.; Barriere, A.; Ausubel, F.M. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol. 2008, 6, 2736–2752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pukkila-Worley, R.; Ausubel, F.M. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr. Opin. Immunol. 2012, 24, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Katz, S.; Huang, H.; Appel, M.D.; Berg, M.; Shapira, M. Association with soil bacteria enhances p38-dependent infection resistance in Caenorhabditis elegans. Infect. Immunity 2013, 81, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Stenuit, B.; Ho, J.; Wang, A.; Parke, C.; Knight, M.; Alvarez-Cohen, L.; Shapira, M. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 2016, 10, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Zhou, X.Y.; Shapira, M. Host-Specific Functional Significance of Caenorhabditis Gut Commensals. Front. Microbiol. 2016, 7, 1622. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, P.; Marsh, S.A.; Braker, I.; Heitland, N.; Wagner, S.; Nakad, R.; Mader, S.; Petersen, C.; Kowallik, V.; Rosenstiel, P.; et al. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biol. 2016, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Rowedder, H.; Braendle, C.; Felix, M.A.; Ruvkun, G. Caenorhabditis elegans responses to bacteria from its natural habitats. Proc. Natl. Acad. Sci. USA 2016, 113, E3941–E3949. [Google Scholar] [CrossRef] [PubMed]
- Rosshart, S.P.; Vassallo, B.G.; Angeletti, D.; Hutchinson, D.S.; Morgan, A.P.; Takeda, K.; Hickman, H.D.; McCulloch, J.A.; Badger, J.H.; Ajami, N.J.; et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 2017, 171, 1015.e13–1028.e13. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.B.; Troemel, E.R. Microbial pathogenesis and host defense in the nematode C. elegans. Curr. Opin. Microbiol. 2015, 23, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.K.; May, R.C. Caenorhabditis elegans, a model organism for investigating immunity. Appl. Environ. Microbiol. 2012, 78, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, I.; Pujol, N. Innate immunity in C. elegans. Adv. Exp. Med. Biol. 2010, 708, 105–121. [Google Scholar] [PubMed]
- Zhang, R.; Hou, A. Host-Microbe Interactions in Caenorhabditis elegans. ISRN Microbiol. 2013, 2013, 356451. [Google Scholar] [CrossRef] [PubMed]
- Ermolaeva, M.A.; Schumacher, B. Insights from the worm: The C. elegans model for innate immunity. Semin. Immunol. 2014, 26, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Miklos, S.; Tan, M.W.; Rahme, L.G.; Ausubel, F.M. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 1999, 96, 47–56. [Google Scholar] [CrossRef]
- Xu, A.; Shi, G.; Liu, F.; Ge, B. Caenorhabditis elegans MOM-4 is required for the activation of the p38 MAPK signaling pathway in the response to Pseudomonas aeruginosa infection. Protein Cell 2013, 4, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Kim, D.H.; Ausubel, F.M. The G protein-coupled receptor FSHR-1 is required for the Caenorhabditis elegans innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 2782–2787. [Google Scholar] [CrossRef] [PubMed]
- Estes, K.A.; Dunbar, T.L.; Powell, J.R.; Ausubel, F.M.; Troemel, E.R. bZIP transcription factor ZIP-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2010, 107, 2153–2158. [Google Scholar] [CrossRef] [PubMed]
- Aballay, A.; Drenkard, E.; Hilbun, L.R.; Ausubel, F.M. Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 2003, 13, 47–52. [Google Scholar] [CrossRef]
- Kurz, C.L.; Chauvet, S.; Andres, E.; Aurouze, M.; Vallet, I.; Michel, G.P.; Uh, M.; Celli, J.; Filloux, A.; De Bentzmann, S.; et al. Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. EMBO J. 2003, 22, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Sifri, C.D.; Begun, J.; Ausubel, F.M.; Calderwood, S.B. Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect. Immunity 2003, 71, 2208–2217. [Google Scholar] [CrossRef]
- Bae, T.; Banger, A.K.; Wallace, A.; Glass, E.M.; Aslund, F.; Schneewind, O.; Missiakas, D.M. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. USA 2004, 101, 12312–12317. [Google Scholar] [CrossRef] [PubMed]
- Bolm, M.; Jansen, W.T.; Schnabel, R.; Chhatwal, G.S. Hydrogen peroxide-mediated killing of Caenorhabditis elegans: A common feature of different streptococcal species. Infect. Immunity 2004, 72, 1192–1194. [Google Scholar] [CrossRef]
- Luallen, R.J.; Bakowski, M.A.; Troemel, E.R. Characterization of microsporidia-induced developmental arrest and a transmembrane leucine-rich repeat protein in Caenorhabditis elegans. PLoS ONE 2015, 10, e0124065. [Google Scholar] [CrossRef] [PubMed]
- Bakowski, M.A.; Desjardins, C.A.; Smelkinson, M.G.; Dunbar, T.A.; Lopez-Moyado, I.F.; Rifkin, S.A.; Cuomo, C.A.; Troemel, E.R. Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLoS Pathog. 2014, 10, e1004200. [Google Scholar] [CrossRef] [PubMed]
- Estes, K.A.; Szumowski, S.C.; Troemel, E.R. Non-lytic, actin-based exit of intracellular parasites from C. elegans intestinal cells. PLoS Pathog. 2011, 7, e1002227. [Google Scholar] [CrossRef] [PubMed]
- Pukkila-Worley, R.; Ausubel, F.M.; Mylonakis, E. Candida albicans infection of Caenorhabditis elegans induces antifungal immune defenses. PLoS Pathog. 2011, 7, e1002074. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Ausubel, F.M.; Perfect, J.R.; Heitman, J.; Calderwood, S.B. Killing of Caenorhabditis elegans by Cryptococcus neoformans as a model of yeast pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 15675–15680. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.J.; Breger, J.; Idnurm, A.; Gerik, K.J.; Lodge, J.K.; Heitman, J.; Calderwood, S.B.; Mylonakis, E. Cryptococcus neoformans gene involved in mammalian pathogenesis identified by a Caenorhabditis elegans progeny-based approach. Infect. Immunity 2005, 73, 8219–8225. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Dishongh, R.; Moore, S.C.; Whitt, M.A.; Chow, M.; Machaca, K. RNA interference is an antiviral defence mechanism in Caenorhabditis elegans. Nature 2005, 436, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Schott, D.H.; Cureton, D.K.; Whelan, S.P.; Hunter, C.P. An antiviral role for the RNA interference machinery in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2005, 102, 18420–18424. [Google Scholar] [CrossRef] [PubMed]
- Gammon, D.B.; Ishidate, T.; Li, L.; Gu, W.; Silverman, N.; Mello, C.C. The Antiviral RNA Interference Response Provides Resistance to Lethal Arbovirus Infection and Vertical Transmission in Caenorhabditis elegans. Curr. Biol. CB 2017, 27, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Lin, Y.L.; Wang, J.P.; Liou, W.; Hou, R.F.; Wu, Y.C.; Liao, C.L. Restriction of vaccinia virus replication by a CED-3 and CED-4-dependent pathway in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2006, 103, 4174–4179. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Maduro, M.; Li, F.; Li, H.W.; Broitman-Maduro, G.; Li, W.X.; Ding, S.W. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature 2005, 436, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.A.; Lee, J.H.; Chapados, B.R.; Debler, E.W.; Schneemann, A.; Williamson, J.R. Dual modes of RNA-silencing suppression by Flock House virus protein B2. Nat. Struct. Mol. Biol. 2005, 12, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Yigit, E.; Li, W.X.; Ding, S.W. An RIG-I-Like RNA helicase mediates antiviral RNAi downstream of viral siRNA biogenesis in Caenorhabditis elegans. PLoS Pathog. 2009, 5, e1000286. [Google Scholar] [CrossRef] [PubMed]
- Felix, M.A.; Ashe, A.; Piffaretti, J.; Wu, G.; Nuez, I.; Belicard, T.; Jiang, Y.; Zhao, G.; Franz, C.J.; Goldstein, L.D.; et al. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol. 2011, 9, e1000586. [Google Scholar] [CrossRef] [PubMed]
- Franz, C.J.; Zhao, G.; Felix, M.A.; Wang, D. Complete genome sequence of Le Blanc virus, a third Caenorhabditis nematode-infecting virus. J. Virol. 2012, 86, 11940. [Google Scholar] [CrossRef] [PubMed]
- Franz, C.J.; Renshaw, H.; Frezal, L.; Jiang, Y.; Felix, M.A.; Wang, D. Orsay, Santeuil and Le Blanc viruses primarily infect intestinal cells in Caenorhabditis nematodes. Virology 2014, 448, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Franz, C.J.; Wu, G.; Renshaw, H.; Zhao, G.; Firth, A.E.; Wang, D. Orsay virus utilizes ribosomal frameshifting to express a novel protein that is incorporated into virions. Virology 2014, 450–451, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Ashe, A.; Belicard, T.; Le Pen, J.; Sarkies, P.; Frezal, L.; Lehrbach, N.J.; Felix, M.A.; Miska, E.A. A deletion polymorphism in the Caenorhabditis elegans RIG-I homolog disables viral RNA dicing and antiviral immunity. eLife 2013, 2, e00994. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, R.; Wang, J.; Ding, S.W.; Lu, R. Homologous RIG-I-like helicase proteins direct RNAi-mediated antiviral immunity in C. elegans by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2013, 110, 16085–16090. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Lu, R. Characterization of virus-encoded RNAi suppressors in Caenorhabditis elegans. J. Virol. 2013, 87, 5414–5423. [Google Scholar] [CrossRef] [PubMed]
- Le Pen, J.; Jiang, H.; Domenico, T.D.; Kneuss, E.; Kosalka, J.; Morgan, M.; Much, M.; Rudolph, L.M.K.; Enright, A.J.; O’Carroll, D.; et al. Terminal uridylyltransferases target RNA viruses as part of the innate immune system in animals. bioRxiv 2017. [Google Scholar] [CrossRef]
- Sarkies, P.; Ashe, A.; Le Pen, J.; McKie, M.A.; Miska, E.A. Competition between virus-derived and endogenous small RNAs regulates gene expression in Caenorhabditis elegans. Genome Res. 2013, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Franz, C.J.; Jiang, H.; Jiang, Y.; Wang, D. An evolutionarily conserved transcriptional response to viral infection in Caenorhabditis nematodes. BMC Genom. 2017, 18, 303. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, M.; Veron, L.; Stempor, P.; Ahringer, J.; Sarkies, P.; Miska, E.A. An Alternative STAT Signaling Pathway Acts in Viral Immunity in Caenorhabditis elegans. mBio 2017, 8, e00924-17. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, K.; Sandoval, L.E.; Leung, C.; Wang, D. An Evolutionarily Conserved Pathway Essential for Orsay Virus Infection of Caenorhabditis elegans. mBio 2017, 8, e00940-17. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.Z.; Hale, K.; Carta, L.; Platzer, E.; Wong, C.; Fang, S.C.; Aroian, R.V. Bacillus thuringiensis crystal proteins that target nematodes. Proc. Natl. Acad. Sci. USA 2003, 100, 2760–2765. [Google Scholar] [CrossRef] [PubMed]
- Los, F.C.; Kao, C.Y.; Smitham, J.; McDonald, K.L.; Ha, C.; Peixoto, C.A.; Aroian, R.V. RAB-5- and RAB-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell Host Microbe 2011, 9, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Spanier, B.; Starke, M.; Higel, F.; Scherer, S.; Fuchs, T.M. Yersinia enterocolitica infection and tcaA-dependent killing of Caenorhabditis elegans. Appl. Environ. Microbiol. 2010, 76, 6277–6285. [Google Scholar] [CrossRef] [PubMed]
- Kothe, M.; Antl, M.; Huber, B.; Stoecker, K.; Ebrecht, D.; Steinmetz, I.; Eberl, L. Killing of Caenorhabditis elegans by Burkholderia cepacia is controlled by the CEP quorum-sensing system. Cell. Microbiol. 2003, 5, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.H.; Chua, K.L.; Chua, H.H.; Liu, B.; Hii, C.S.; Chong, H.L.; Tan, P. Characterization of Burkholderia pseudomallei infection and identification of novel virulence factors using a Caenorhabditis elegans host system. Mol. Microbiol. 2002, 44, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Garsin, D.A.; Sifri, C.D.; Mylonakis, E.; Qin, X.; Singh, K.V.; Murray, B.E.; Calderwood, S.B.; Ausubel, F.M. A simple model host for identifying Gram-positive virulence factors. Proc. Natl. Acad. Sci. USA 2001, 98, 10892–10897. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, L.E.; Slutz, S.S.; Tan, M.W.; Ingmer, H. Caenorhabditis elegans is a model host for Listeria monocytogenes. Appl. Environ. Microbiol. 2006, 72, 1700–1701. [Google Scholar] [CrossRef] [PubMed]
- Pradel, E.; Zhang, Y.; Pujol, N.; Matsuyama, T.; Bargmann, C.I.; Ewbank, J.J. Detection and avoidance of a natural product from the pathogenic bacterium Serratia marcescens by Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, H.; Ewbank, J.J. The genetics of pathogen avoidance in Caenorhabditis elegans. Mol. Microbiol. 2007, 66, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, H.; Kurz, C.L.; Ewbank, J.J. Evolution of the innate immune system: The worm perspective. Immunol. Rev. 2004, 198, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Darby, C. Interactions with microbial pathogens. In WormBook: The Online Review of C. elegans Biology; The C. elegans Research Community: Birmingham, AL, USA, 2005; pp. 1–15. [Google Scholar]
- Gordon, S. Pattern recognition receptors: Doubling up for the innate immune response. Cell 2002, 111, 927–930. [Google Scholar] [CrossRef]
- Nicholas, H.R.; Hodgkin, J. Responses to infection and possible recognition strategies in the innate immune system of Caenorhabditis elegans. Mol. Immunol. 2004, 41, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Rajamuthiah, R.; Mylonakis, E. Effector triggered immunity. Virulence 2014, 5, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Feinbaum, R.; Alloing, G.; Emerson, F.E.; Garsin, D.A.; Inoue, H.; Tanaka-Hino, M.; Hisamoto, N.; Matsumoto, K.; Tan, M.W.; et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 2002, 297, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Bolz, D.D.; Tenor, J.L.; Aballay, A. A conserved PMK-1/p38 MAPK is required in Caenorhabditis elegans tissue-specific immune response to Yersinia pestis infection. J. Biol. Chem. 2010, 285, 10832–10840. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; van der Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 2004, 101, 10995–11000. [Google Scholar] [CrossRef] [PubMed]
- Troemel, E.R.; Chu, S.W.; Reinke, V.; Lee, S.S.; Ausubel, F.M.; Kim, D.H. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genetics 2006, 2, e183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, E.A.; Chen, W.C.; Tan, M.W. The DAF-2 insulin-like signaling pathway independently regulates aging and immunity in C. elegans. Aging Cell 2008, 7, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Aballay, A. Regulation of DAF-16-mediated Innate Immunity in Caenorhabditis elegans. J. Biol. Chem. 2009, 284, 35580–35587. [Google Scholar] [CrossRef] [PubMed]
- Shapira, M.; Hamlin, B.J.; Rong, J.; Chen, K.; Ronen, M.; Tan, M.W. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 14086–14091. [Google Scholar] [CrossRef] [PubMed]
- Visvikis, O.; Ihuegbu, N.; Labed, S.A.; Luhachack, L.G.; Alves, A.F.; Wollenberg, A.C.; Stuart, L.M.; Stormo, G.D.; Irazoqui, J.E. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 2014, 40, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Jansson, H.B. Adhesion of Conidia of Drechmeria coniospora to Caenorhabditis elegans Wild Type and Mutants. J. Nematol. 1994, 26, 430–435. [Google Scholar] [PubMed]
- Zugasti, O.; Bose, N.; Squiban, B.; Belougne, J.; Kurz, C.L.; Schroeder, F.C.; Pujol, N.; Ewbank, J.J. Activation of a G protein-coupled receptor by its endogenous ligand triggers the innate immune response of Caenorhabditis elegans. Nat. Immunol. 2014, 15, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Diogo, J.; Bratanich, A. The nematode Caenorhabditis elegans as a model to study viruses. Arch. Virol. 2014, 159, 2843–2851. [Google Scholar] [CrossRef] [PubMed]
- Gammon, D.B. Caenorhabditis elegans as an Emerging Model for Virus-Host Interactions. J. Virol. 2017, 91, e00509-17. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.; Estevez, A.; Yin, X.; Fox, R.; Morrison, R.; McDonnell, M.; Gleason, C.; Miller, D.M.; Strange, K. A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 2002, 33, 503–514. [Google Scholar] [CrossRef]
- Zhang, S.; Banerjee, D.; Kuhn, J.R. Isolation and culture of larval cells from C. elegans. PLoS ONE 2011, 6, e19505. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Diaz, E.; Stefanakis, N.; Carrera, I.; Glenwinkel, L.; Wang, G.; Driscoll, M.; Hobert, O. Silencing of Repetitive DNA Is Controlled by a Member of an Unusual Caenorhabditis elegans Gene Family. Genetics 2017, 207, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.H. Adaptive evolution in two large families of ubiquitin-ligase adapters in nematodes and plants. Genome Res. 2006, 16, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.C.; Dror, T.; Sowa, J.N.; Panek, J.; Chen, K.; Lim, E.S.; Wang, D.; Troemel, E.R. An Intracellular Pathogen Response Pathway Promotes Proteostasis in C. elegans. Curr. Biol. CB 2017, 27, 3544.e5–3553.e5. [Google Scholar] [CrossRef] [PubMed]
- Jose, A.M.; Kim, Y.A.; Leal-Ekman, S.; Hunter, C.P. Conserved tyrosine kinase promotes the import of silencing RNA into Caenorhabditis elegans cells. Proc. Natl. Acad. Sci. USA 2012, 109, 14520–14525. [Google Scholar] [CrossRef] [PubMed]
- Tampakakis, E.; Peleg, A.Y.; Mylonakis, E. Interaction of Candida albicans with an intestinal pathogen, Salmonella enterica serovar Typhimurium. Eukaryot. Cell 2009, 8, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; Tampakakis, E.; Fuchs, B.B.; Eliopoulos, G.M.; Moellering, R.C., Jr.; Mylonakis, E. Prokaryote-eukaryote interactions identified by using Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2008, 105, 14585–14590. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Kingdoms | Pathogens | Infection and Pathogenic Mode | Host Pathway and Response in the Intestine | Reference(s) |
|---|---|---|---|---|
| Bacteria | P. aeruginosa | Feeding; slow killing by colonization and fast killing by toxin | PMK-1, ZIP-2, FSHR-1 dependent pathways | [19,20,21,22] |
| S. enterica | Feeding; killing by colonization; LPS as virulence factor | PMK-1-dependant programmed cell death pathway | [23] | |
| S. marcescens | Feeding; colonization causes distended intestine; LPS and hemolysin as virulence factors | DBL-1/TGF-β pathway | [24] | |
| S. aureus | Feeding; α-hemolysin as virulence factor | SEK-1 and NSY-1 dependent p38 MAP kinase pathway and TFEB mediated transcriptional response | [25,26] | |
| S. pyogenes | Feeding; colonization; hydrogen peroxide as virulence factor | Not analyzed | [27] | |
| Fungi | N. parisii | Feeding; intracellular infection | CUL-6, SKR-3, 4, 5 ubiquitin ligase pathways | [6,28,29,30] |
| C. albicans | Feeding; colonization; | PMK-1/p-38 MAPK pathways | [31] | |
| C. neoformans | Feeding; colonization; laccase; polysaccharide capsule and/or melanization as virulence factors | CED-1, C03F11.3 and ABL-1 dependent pathways | [32,33] | |
| Viruses | VSV | Infection of primary cell culture or microinjection | RNA interference | [34,35,36] |
| Vaccinia virus | PEG permeabilization | CED-3 and CED-4 Cell death pathways | [37] | |
| Flock house virus | Transgenic initiation of virus replication | RNA interference | [38,39,40] | |
| Orsay virus | Feeding | RNA interference CUL-6 ubiquitin-proteasome degradation; STA-1 repression and CDE-1 | [41,42,43,44,45,46,47,48,49,50,51,52] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Wang, D. The Microbial Zoo in the C. elegans Intestine: Bacteria, Fungi and Viruses. Viruses 2018, 10, 85. https://doi.org/10.3390/v10020085
Jiang H, Wang D. The Microbial Zoo in the C. elegans Intestine: Bacteria, Fungi and Viruses. Viruses. 2018; 10(2):85. https://doi.org/10.3390/v10020085
Chicago/Turabian StyleJiang, Hongbing, and David Wang. 2018. "The Microbial Zoo in the C. elegans Intestine: Bacteria, Fungi and Viruses" Viruses 10, no. 2: 85. https://doi.org/10.3390/v10020085
APA StyleJiang, H., & Wang, D. (2018). The Microbial Zoo in the C. elegans Intestine: Bacteria, Fungi and Viruses. Viruses, 10(2), 85. https://doi.org/10.3390/v10020085