Pharmacological Inhibition of Protein Kinase C Reduces West Nile Virus Replication




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Drug Treatments
2.3. Quantitative RT-PCR
2.4. Antibodies
2.5. Immunofluorescence
2.6. Western Blot
2.7. Statistical Analyses
3. Results
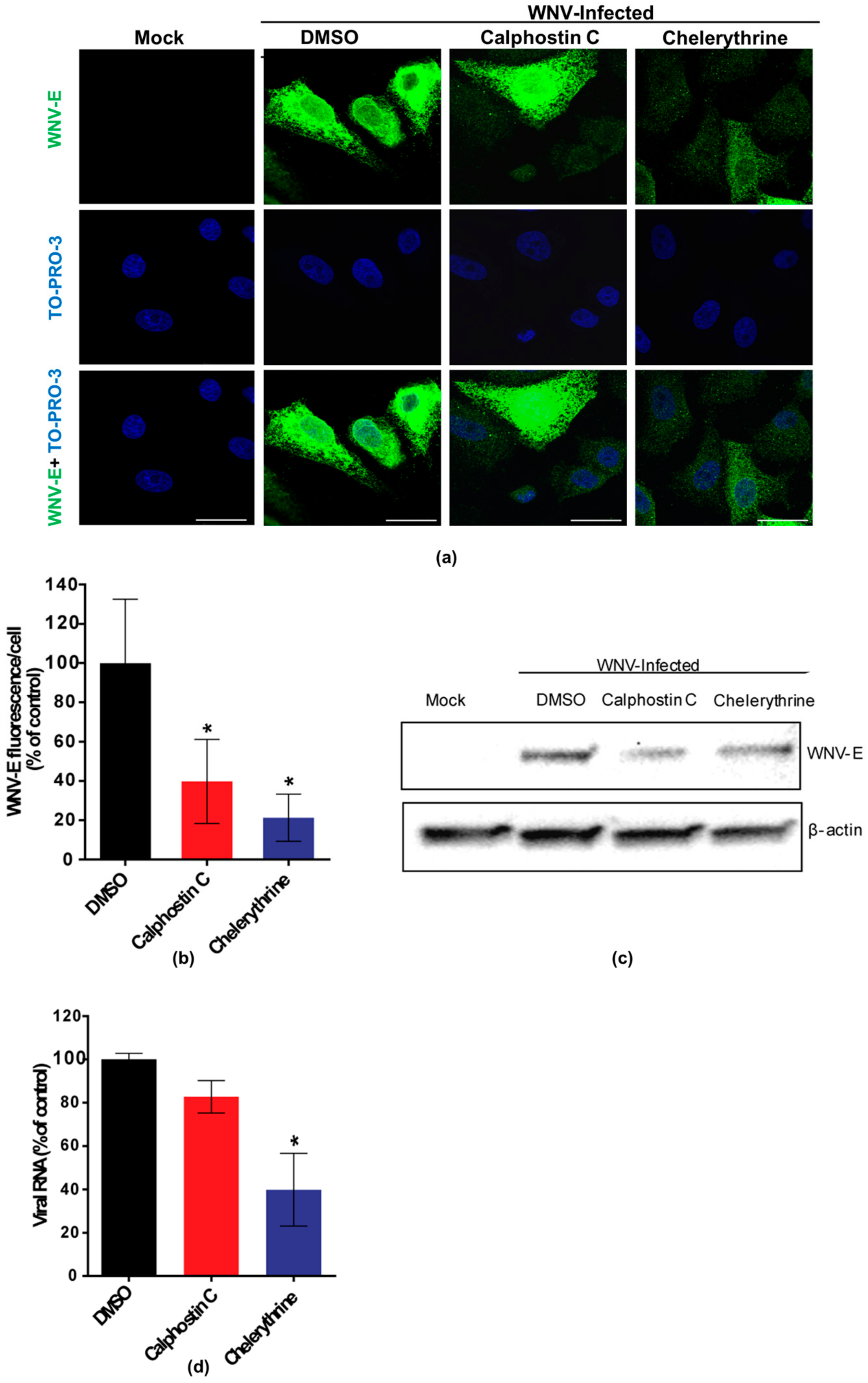
3.1. Inhibition of WNV Multiplication after Treatment with PKC Inhibitors
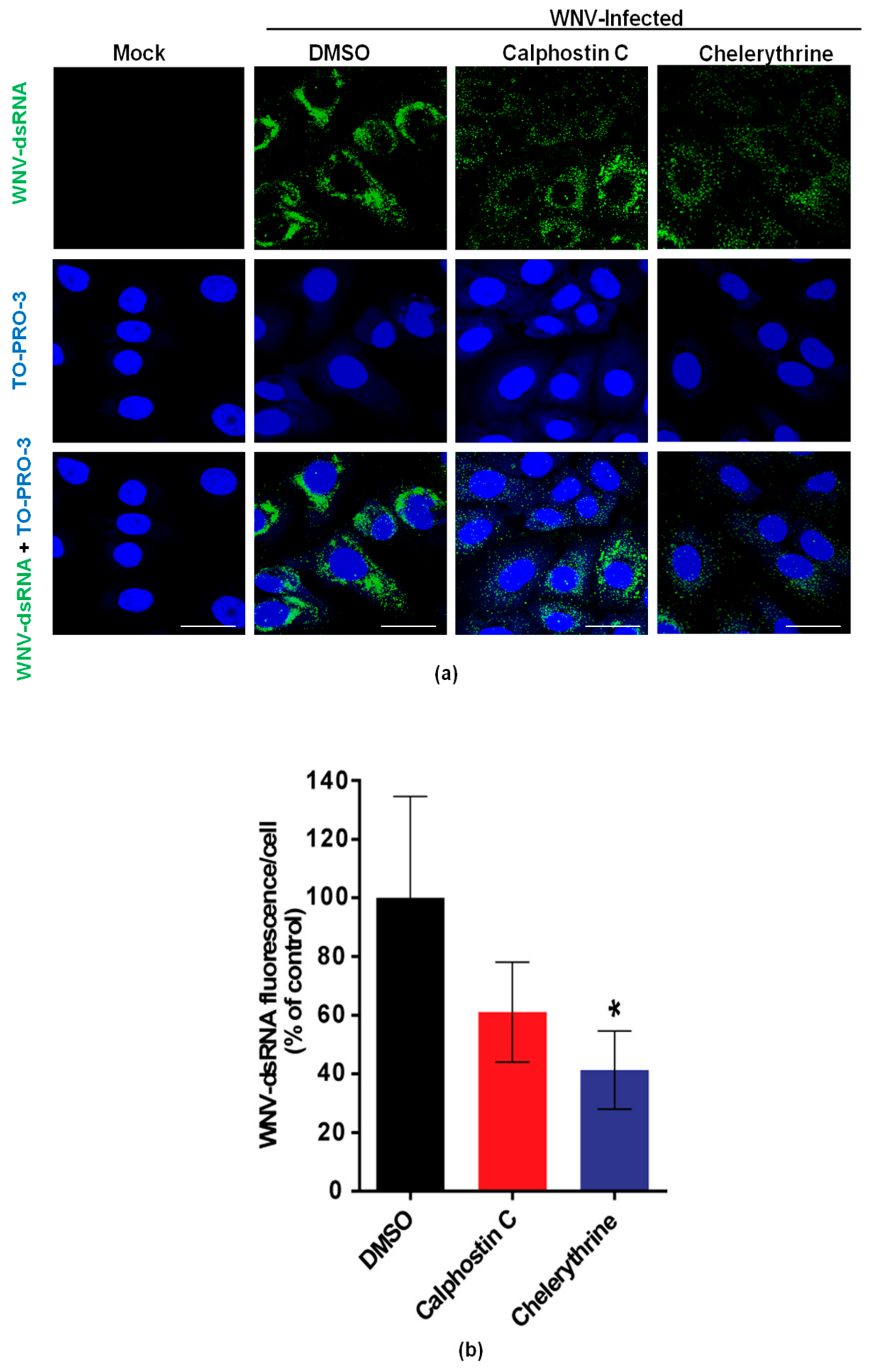
3.2. Chelerythrine Inhibition Reduces WNV Infection at a Replication Step
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martin-Acebes, M.A.; Saiz, J.C. West nile virus: A re-emerging pathogen revisited. World J. Virol. 2012, 1, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Martin-Acebes, M.A.; Blazquez, A.B.; Jimenez de Oya, N.; Escribano-Romero, E.; Saiz, J.C. West nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS ONE 2011, 6, e24970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Brodie, C.; Blumberg, P.M. Regulation of cell apoptosis by protein kinase c delta. Apoptosis 2003, 8, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. Pkc and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Sontag, J.M.; Garcia, A. Protein phosphatase 2a is a critical regulator of protein kinase c zeta signaling targeted by sv40 small t to promote cell growth and nf-kappab activation. EMBO J. 1997, 16, 5662–5671. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Structural basis of protein kinase c isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Way, K.J.; Chou, E.; King, G.L. Identification of pkc-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol. Sci. 2000, 21, 181–187. [Google Scholar] [CrossRef]
- Webb, B.L.; Hirst, S.J.; Giembycz, M.A. Protein kinase c isoenzymes: A review of their structure, regulation and role in regulating airways smooth muscle tone and mitogenesis. Br. J. Pharmacol. 2000, 130, 1433–1452. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, W.S.; Apostolatos, A.H.; Ostrov, D.A.; Acevedo-Duncan, M. Two novel atypical pkc inhibitors; acpd and dnda effectively mitigate cell proliferation and epithelial to mesenchymal transition of metastatic melanoma while inducing apoptosis. Int. J. Oncol. 2017, 51, 1370–1382. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. The role of protein kinase c in cell surface signal transduction and tumour promotion. Nature 1984, 308, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Sobhia, M.E.; Grewal, B.K.; Ml, S.P.; Patel, J.; Kaur, A.; Haokip, T.; Kokkula, A. Protein kinase c inhibitors: A patent review (2008–2009). Expert Opin. Ther. Pat. 2013, 23, 1297–1315. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Dawson, A.R.; Potts, G.K.; Freiberger, E.C.; Baker, S.F.; Moser, L.A.; Bernard, K.A.; Coon, J.J.; Mehle, A. Influenza virus recruits host protein kinase c to control assembly and activity of its replication machinery. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Debing, Y.; Zhou, X.; Yin, Y.; Xu, L.; Herrera Carrillo, E.; Brandsma, J.H.; Poot, R.A.; Berkhout, B.; et al. Biological or pharmacological activation of protein kinase c alpha constrains hepatitis e virus replication. Antivir. Res 2017, 140, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, A.; Takahama, S.; Sawasaki, T.; Ode, H.; Yokoyama, M.; Okayama, A.; Ishikawa, A.; Miyakawa, K.; Matsunaga, S.; Kimura, H.; et al. The phosphorylation of hiv-1 gag by atypical protein kinase c facilitates viral infectivity by promoting vpr incorporation into virions. Retrovirology 2014, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Nuesch, J.P.; Christensen, J.; Rommelaere, J. Initiation of minute virus of mice DNA replication is regulated at the level of origin unwinding by atypical protein kinase c phosphorylation of ns1. J. Virol. 2001, 75, 5730–5739. [Google Scholar] [CrossRef] [PubMed]
- Noppakunmongkolchai, W.; Poyomtip, T.; Jittawuttipoka, T.; Luplertlop, N.; Sakuntabhai, A.; Chimnaronk, S.; Jirawatnotai, S.; Tohtong, R. Inhibition of protein kinase c promotes dengue virus replication. Virol. J. 2016, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Hoover, S.; Falk, S.P.; Weisblum, B.; Vestling, M.; Striker, R. Phosphorylation of yellow fever virus ns5 alters methyltransferase activity. Virology 2008, 380, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.A.; Bhattacharya, D.; Lim, P.Y.; Falk, S.; Weisblum, B.; Bernard, K.A.; Sharma, M.; Kuhn, R.J.; Striker, R. West nile virus methyltransferase domain interacts with protein kinase g. Virol. J. 2013, 10, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, J.M.; Kenney, M.T.; Westaway, E.G. West nile virus strain kunjin ns5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral rna synthesis. J. Gen. Virol. 2007, 88, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.V.; Tsekhanovskaya, N.A.; Maksimova, T.G.; Bachvalova, V.N.; Matveeva, V.A.; Kit, Y. Phosphorylation of tick-borne encephalitis virus ns5 protein. Virus Res. 1997, 49, 9–15. [Google Scholar] [CrossRef]
- Reed, K.E.; Gorbalenya, A.E.; Rice, C.M. The ns5a/ns5 proteins of viruses from three genera of the family flaviviridae are phosphorylated by associated serine/threonine kinases. J. Virol. 1998, 72, 6199–6206. [Google Scholar] [PubMed]
- Xiao, H.; Liu, M. Atypical protein kinase c in cell motility. Cell Mol. Life Sci. 2012, 70, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Martin-Acebes, M.A.; Merino-Ramos, T.; Blazquez, A.B.; Casas, J.; Escribano-Romero, E.; Sobrino, F.; Saiz, J.C. The composition of west nile virus lipid envelope unveils a role of sphingolipid metabolism in flavivirus biogenesis. J. Virol. 2014, 88, 12041–12054. [Google Scholar] [CrossRef] [PubMed]
- Bhuvanakantham, R.; Cheong, Y.K.; Ng, M.L. West nile virus capsid protein interaction with importin and hdm2 protein is regulated by protein kinase c-mediated phosphorylation. Microbes Infect. 2010, 12, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.J.; Leong, P.W.; Ng, M.L. Analysis of the endocytic pathway mediating the infectious entry of mosquito-borne flavivirus west nile into aedes albopictus mosquito (c6/36) cells. Virology 2006, 349, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V.; Lee, M.C.; Sajan, M.P. Atypical pkc: A target for treating insulin-resistant disorders of obesity, the metabolic syndrome and type 2 diabetes mellitus. Expert Opin. Ther. Targets 2014, 18, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, M.D.; Krull, J.E.; Scarlett, J.M.; Guyenet, S.J.; Sajan, M.P.; Damian, V.; Nguyen, H.T.; Leitges, M.; Morton, G.J.; Farese, R.V.; et al. Deletion of protein kinase c lambda in pomc neurons predisposes to diet-induced obesity. Diabetes 2017, 66, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, X.; Wang, H.; Zhao, B.; Wu, X.; Su, L.; Xie, S.; Wang, Y.; Li, J.; Liu, J.; et al. Pkczeta as a promising therapeutic target for tnfalpha-induced inflammatory disorders in chronic cutaneous wounds. Int. J. Mol. Med. 2017, 40, 1335–1346. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Martin-Acebes, M.A.; Saiz, J.C. Inhibition of west nile virus multiplication in cell culture by anti-parkinsonian drugs. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Kerst, A.J. Nucleic acid sequence-based amplification assays for rapid detection of west nile and st. Louis encephalitis viruses. J. Clin. Microbiol. 2001, 39, 4506–4513. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Saiz, J.C. West nile virus (wnv) transmission routes in the murine model: Intrauterine, by breastfeeding and after cannibal ingestion. Virus Res. 2010, 151, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Romero, E.; Gamino, V.; Merino-Ramos, T.; Blazquez, A.B.; Martin-Acebes, M.A.; de Oya, N.J.; Gutierrez-Guzman, A.V.; Escribano, J.M.; Hofle, U.; Saiz, J.C. Protection of red-legged partridges (alectoris rufa) against west nile virus (wnv) infection after immunization with wnv recombinant envelope protein e (re). Vaccine 2013, 31, 4523–4527. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martin-Acebes, M.A. Infection with usutu virus induces an autophagic response in mammalian cells. PLoS Negl. Trop Dis. 2013, 7, e2509. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The protein kinase cbeta-selective inhibitor, enzastaurin (ly317615.Hcl), suppresses signaling through the akt pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Ringvold, H.C.; Khalil, R.A. Protein kinase c as regulator of vascular smooth muscle function and potential target in vascular disorders. Adv. Pharmacol. 2017, 78, 203–301. [Google Scholar] [PubMed]
- Herbert, J.M.; Augereau, J.M.; Gleye, J.; Maffrand, J.P. Chelerythrine is a potent and specific inhibitor of protein kinase c. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef]
- Boldescu, V.; Behnam, M.A.M.; Vasilakis, N.; Klein, C.D. Broad-spectrum agents for flaviviral infections: Dengue, zika and beyond. Nat. Rev. Drug Discov. 2017, 16, 565–586. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.L.; Padilla, L.; Castano, J.C. Inhibitors compounds of the flavivirus replication process. Virol. J. 2017, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Saiz, J.C.; Martin-Acebes, M.A. The race to find antivirals for zika virus. Antimicrob. Agents Chemother. 2017, 61, e00411-17. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Kong, C.; Zhang, Z.; Zhu, Y.; Zhang, Y.; Chen, X. Reduction of protein kinase c alpha (pkc-alpha) promote apoptosis via down-regulation of dicer in bladder cancer. J. Cell Mol. Med. 2015, 19, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Zhan, B.; Kong, C.; Guo, K.; Zhang, Z. Pkcalpha is involved in the progression of kidney carcinoma through regulating netrin-1/unc5b signaling pathway. Tumour. Biol. 2013, 34, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pan, Y.; Zhang, G.; Wu, Y.; Zhong, W.; Chu, C.; Qian, Y.; Zhu, G. Chelerythrine inhibits human hepatocellular carcinoma metastasis in vitro. Biol. Pharm. Bull. 2017, 41, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Yamboliev, I.A.; Mutafova-Yambolieva, V.N. Pi3k and pkc contribute to membrane depolarization mediated by alpha2-adrenoceptors in the canine isolated mesenteric vein. BMC Physiol. 2005, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- De, B.P.; Gupta, S.; Banerjee, A.K. Cellular protein kinase c isoform zeta regulates human parainfluenza virus type 3 replication. Proc. Natl. Acad. Sci. USA 1995, 92, 5204–5208. [Google Scholar] [CrossRef] [PubMed]
- Filone, C.M.; Hanna, S.L.; Caino, M.C.; Bambina, S.; Doms, R.W.; Cherry, S. Rift valley fever virus infection of human cells and insect hosts is promoted by protein kinase c epsilon. PLoS ONE 2010, 5, e15483. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Ennaciri, J.; Cordeiro, P.; El Bassam, S.; Menezes, J. Herpes simplex virus-1 up-regulates il-15 gene expression in monocytic cells through the activation of protein tyrosine kinase and pkc zeta/lambda signaling pathways. J. Mol. Biol. 2007, 367, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.; Staber, J.; Thomas, K.; Hunninghake, G. Respiratory syncytial virus infection results in activation of multiple protein kinase c isoforms leading to activation of mitogen-activated protein kinase. J. Immunol. 2001, 166, 2681–2687. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Akula, S.M.; Zien, C.A.; Krishnan, H.H.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-pkc-zeta-mek-erk signaling pathway in target cells early during infection: Implications for infectivity. J. Virol. 2003, 77, 1524–1539. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blázquez, A.B.; Vázquez-Calvo, Á.; Martín-Acebes, M.A.; Saiz, J.-C. Pharmacological Inhibition of Protein Kinase C Reduces West Nile Virus Replication. Viruses 2018, 10, 91. https://doi.org/10.3390/v10020091
Blázquez AB, Vázquez-Calvo Á, Martín-Acebes MA, Saiz J-C. Pharmacological Inhibition of Protein Kinase C Reduces West Nile Virus Replication. Viruses. 2018; 10(2):91. https://doi.org/10.3390/v10020091
Chicago/Turabian StyleBlázquez, Ana B., Ángela Vázquez-Calvo, Miguel A. Martín-Acebes, and Juan-Carlos Saiz. 2018. "Pharmacological Inhibition of Protein Kinase C Reduces West Nile Virus Replication" Viruses 10, no. 2: 91. https://doi.org/10.3390/v10020091
APA StyleBlázquez, A. B., Vázquez-Calvo, Á., Martín-Acebes, M. A., & Saiz, J.-C. (2018). Pharmacological Inhibition of Protein Kinase C Reduces West Nile Virus Replication. Viruses, 10(2), 91. https://doi.org/10.3390/v10020091