Are microRNAs Important Players in HIV-1 Infection? An Update

Abstract
:1. Introduction
2. HIV-1 Replication
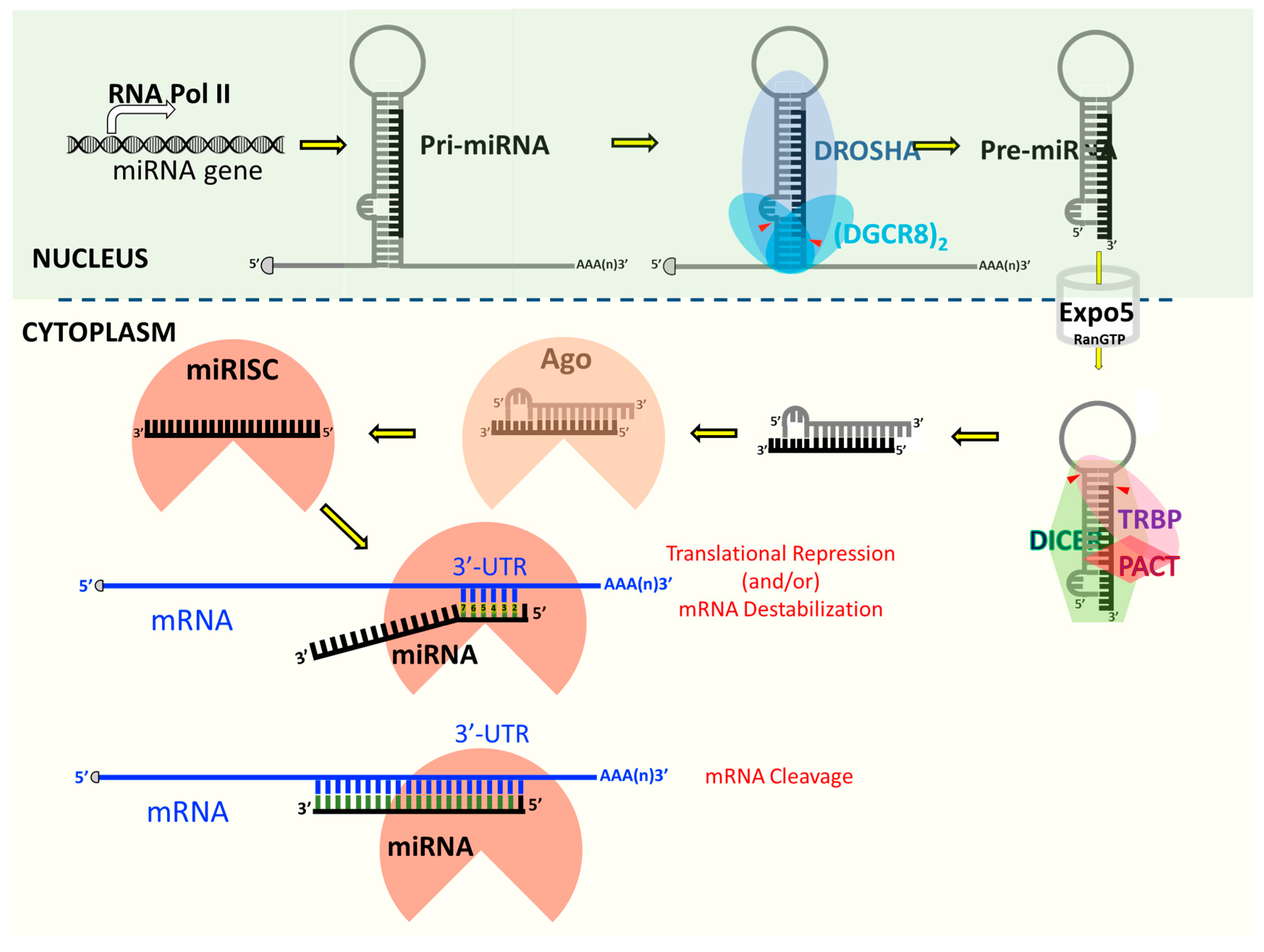
3. Regulation of Cellular Gene Expression by Small Non-Coding RNAs
4. Cellular microRNAs
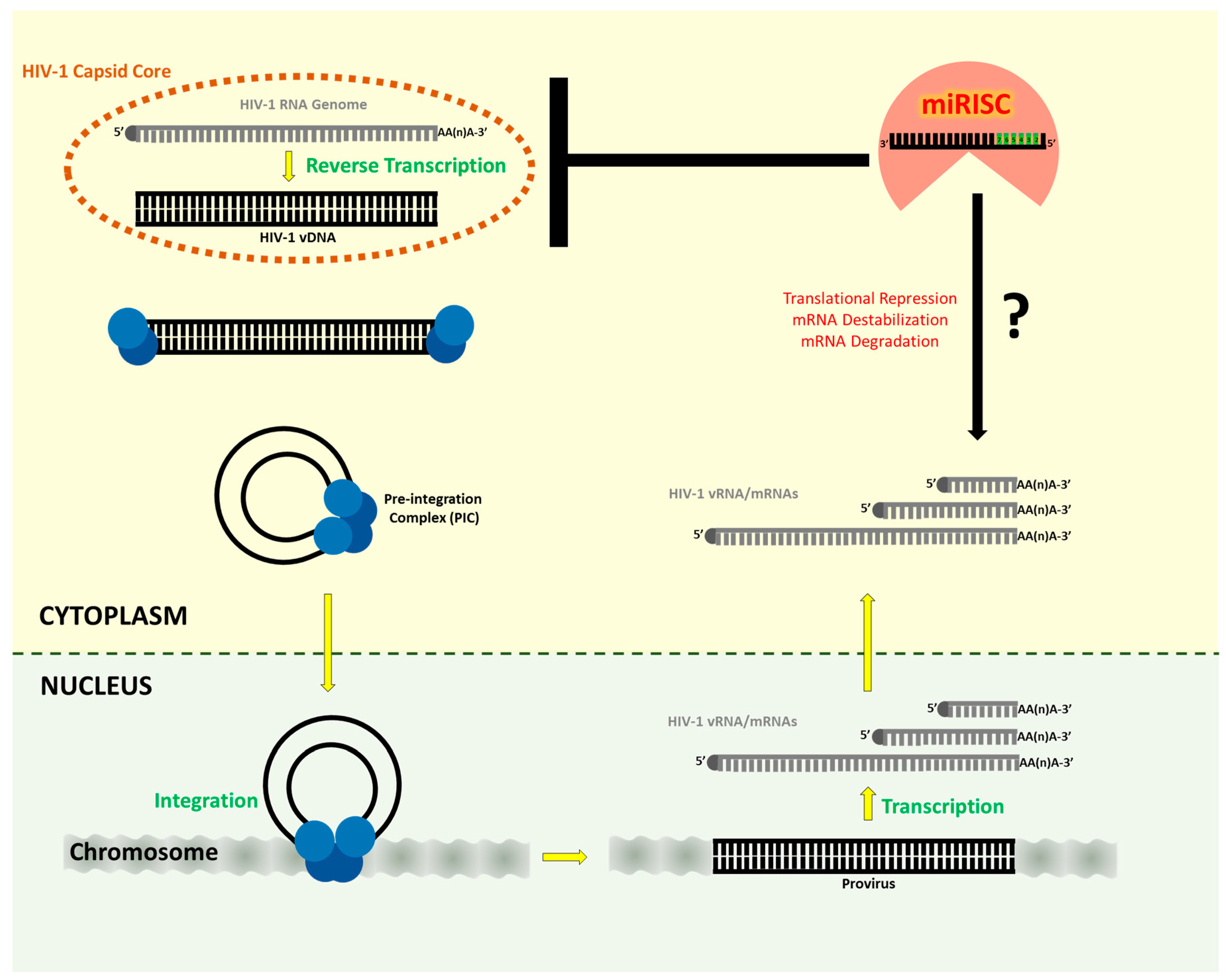
5. Cellular miRNAs as Cofactors in HIV-1 Replication
6. Cellular miRNAs as Anti-Viral Factors in HIV-1 Replication
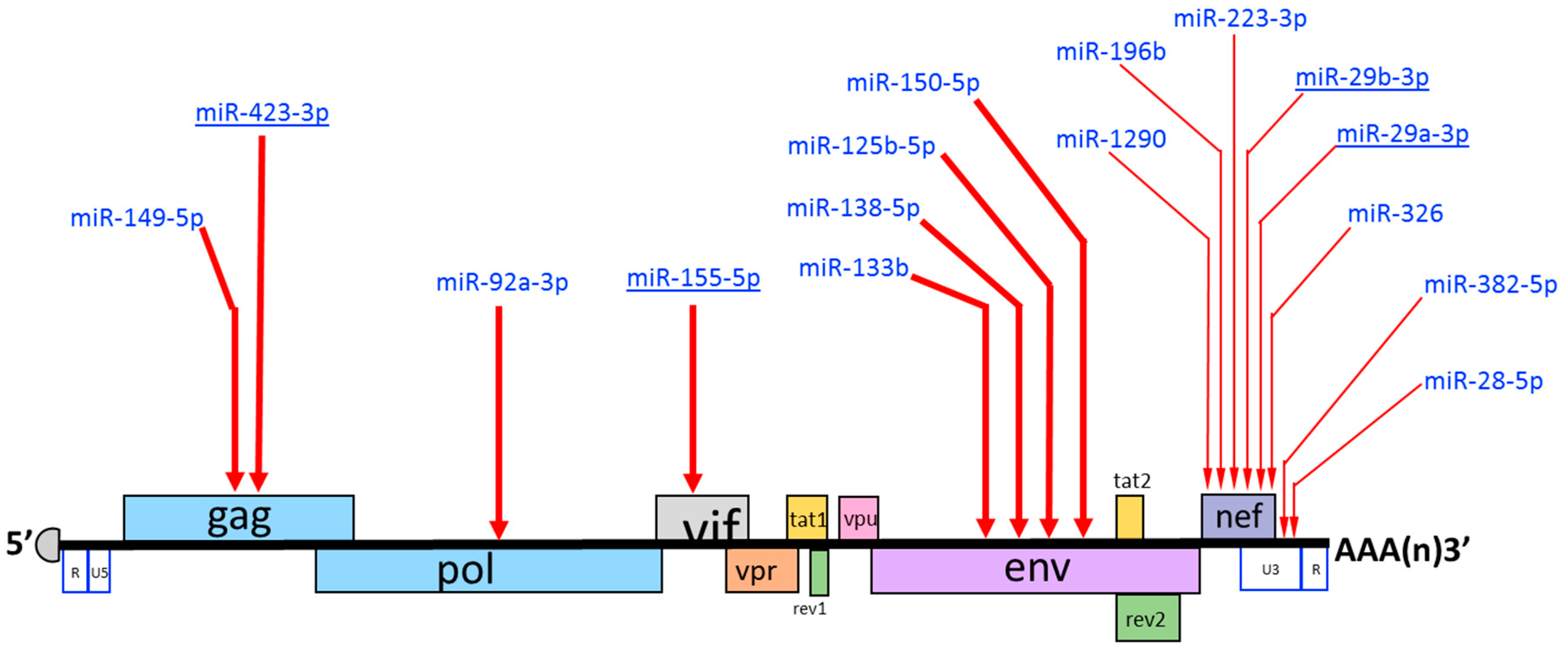
7. Does HIV-1 Encode miRNAs?
8. Concluding Remarks and Outstanding Questions
Acknowledgments
Conflicts of Interest
References
- Swanstrom, R.; Coffin, J. HIV-1 pathogenesis: The virus. Cold Spring Harb. Perspect. Med. 2012, 2, a007443. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; McMichael, A. The T-cell response to HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007054. [Google Scholar] [CrossRef] [PubMed]
- Overbaugh, J.; Morris, L. The Antibody Response against HIV-1. Cold Spring Harb. Perspect. Med. 2012, 2, a007039. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pepin, J.; et al. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Watts, T.D.; McKay, R.A.; Suchard, M.A.; Granade, T.; Teuwen, D.E.; Koblin, B.A.; Heneine, W.; Lemey, P.; Jaffe, H.W. 1970s and ‘Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 2016, 539, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Picker, L.J.; Hansen, S.G.; Lifson, J.D. New paradigms for HIV/AIDS vaccine development. Annu. Rev. Med. 2012, 63, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.E.; D’Couto, H.T.; Barouch, D.H. New concepts in HIV-1 vaccine development. Curr. Opin. Immunol. 2016, 41, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Burton, D.R. Developing an HIV vaccine. Science 2017, 355, 1129–1130. [Google Scholar] [CrossRef] [PubMed]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Geller, R.; Garijo, R.; Lopez-Aldeguer, J.; Sanjuan, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Laskey, S.B.; Siliciano, R.F. A mechanistic theory to explain the efficacy of antiretroviral therapy. Nat. Rev. Microbiol. 2014, 12, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef] [PubMed]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Trono, D.; Van Lint, C.; Rouzioux, C.; Verdin, E.; Barre-Sinoussi, F.; Chun, T.W.; Chomont, N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 2010, 329, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Barre-Sinoussi, F.; Ross, A.L.; Delfraissy, J.F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Jordan, M.R.; Sultan, B.J.; Hill, A.; Davis, D.H.; Gregson, J.; Sawyer, A.W.; Hamers, R.L.; Ndembi, N.; Pillay, D.; et al. Global trends in antiretroviral resistance in treatment-naive individuals with HIV after rollout of antiretroviral treatment in resource-limited settings: A global collaborative study and meta-regression analysis. Lancet 2012, 380, 1250–1258. [Google Scholar] [CrossRef]
- Stadeli, K.M.; Richman, D.D. Rates of emergence of HIV drug resistance in resource-limited settings: A systematic review. Antivir. Ther. 2013, 18, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 drug resistance and resistance testing. Infect Genet. Evol. 2016, 46, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Gregson, J.; Parkin, N.; Haile-Selassie, H.; Tanuri, A.; Andrade Forero, L.; Kaleebu, P.; Watera, C.; Aghokeng, A.; Mutenda, N.; et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: A systematic review and meta-regression analysis. Lancet Infect. Dis. 2017, 18, 346–355. [Google Scholar] [CrossRef]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.A.; Bailey, J.R.; Blankson, J.N. Elucidating the elite: Mechanisms of control in HIV-1 infection. Trends Pharmacol. Sci. 2009, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.; Swanstrom, R. HIV pathogenesis: Dynamics and genetics of viral populations and infected cells. Cold Spring Harb. Perspect. Med. 2013, 3, a012526. [Google Scholar] [CrossRef] [PubMed]
- Lackner, A.A.; Lederman, M.M.; Rodriguez, B. HIV pathogenesis: The host. Cold Spring Harb. Perspect. Med. 2012, 2, a007005. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A.; Johnson, W.E. Host genes important to HIV replication and evolution. Cold Spring Harb. Perspect. Med. 2012, 2, a007203. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; Alter, G. Innate immune control of HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007070. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.; Lythgoe, K.; Leventhal, G.E.; Shirreff, G.; Hollingsworth, T.D.; Alizon, S.; Bonhoeffer, S. Virulence and pathogenesis of HIV-1 infection: An evolutionary perspective. Science 2014, 343, 1243727. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Bloch, N.; Landau, N.R. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Boritz, E.A.; Darko, S.; Swaszek, L.; Wolf, G.; Wells, D.; Wu, X.; Henry, A.R.; Laboune, F.; Hu, J.; Ambrozak, D.; et al. Multiple Origins of Virus Persistence during Natural Control of HIV Infection. Cell 2016, 166, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Hoxie, J.A.; June, C.H. Novel cell and gene therapies for HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007179. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Shaw, G.M.; Korber, B.; Kelsoe, G.; Sodroski, J.; Hahn, B.H.; Borrow, P.; McMichael, A.J. HIV-Host Interactions: Implications for Vaccine Design. Cell Host Microbe 2016, 19, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev. 2011, 25, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Rice, A.P. Mini ways to stop a virus: MicroRNAs and HIV-1 replication. Future Virol. 2011, 6, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Klase, Z.; Houzet, L.; Jeang, K.T. MicroRNAs and HIV-1: Complex interactions. J. Biol. Chem. 2012, 287, 40884–40890. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Murray, D.D.; Kelleher, A.D. miRNAs and HIV: Unforeseen determinants of host-pathogen interaction. Immunol. Rev. 2013, 254, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Navas-Martin, S.; Martin-Garcia, J. MicroRNAs and HIV-1 infection: Antiviral activities and beyond. J. Mol. Biol. 2014, 426, 1178–1197. [Google Scholar] [CrossRef] [PubMed]
- tenOever, B.R. RNA viruses and the host microRNA machinery. Nat. Rev. Microbiol. 2013, 11, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Steitz, J.A. Virus meets host microRNA: The destroyer, the booster, the hijacker. Mol. Cell. Biol. 2014, 34, 3780–3787. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Klimstra, W.B. MicroRNA Regulation of RNA Virus Replication and Pathogenesis. Trends Mol. Med. 2017, 23, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Das, A.T.; Berkhout, B. HIV-1 RNAs: Sense and antisense, large mRNAs and small siRNAs and miRNAs. Curr. Opin. HIV AIDS 2015, 10, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O.; Martin, M.A. Human immundeficiency viruses: Replication. In Fields Virology, 6th ed.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E. HIV-1 biology at the protein level. In HIV-1 Proteomics: From Discovery to Clinical Application; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D’Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; Konig, R.; et al. Host cell factors in HIV replication: Meta-analysis of genome-wide studies. PLoS Pathog. 2009, 5, e1000437. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Engelman, A.N. Capsid-Dependent Host Factors in HIV-1 Infection. Trends Microbiol. 2017, 25, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Gres, A.T.; Kirby, K.A.; KewalRamani, V.N.; Tanner, J.J.; Pornillos, O.; Sarafianos, S.G. STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Perilla, J.R.; Gronenborn, A.M. Molecular Architecture of the Retroviral Capsid. Trends Biochem. Sci. 2016, 41, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Glass, B.; Hagen, W.J.; Krausslich, H.G.; Briggs, J.A. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 2016, 354, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Fassati, A. Multiple roles of the capsid protein in the early steps of HIV-1 infection. Virus Res. 2012, 170, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, Z.; Aiken, C. HIV-1 uncoating: Connection to nuclear entry and regulation by host proteins. Virology 2014, 454–455, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R.; Bushman, F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Engelman, A. Viral and cellular requirements for the nuclear entry of retroviral preintegration nucleoprotein complexes. Viruses 2013, 5, 2483–2511. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Karn, J.; Stoltzfus, C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012, 2, a006916. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Krausslich, H.G. The molecular architecture of HIV. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Yeager, M.; Pornillos, O. Assembly and architecture of HIV. Adv. Exp. Med. Biol. 2012, 726, 441–465. [Google Scholar] [PubMed]
- Schur, F.K.; Hagen, W.J.; Rumlova, M.; Ruml, T.; Muller, B.; Krausslich, H.G.; Briggs, J.A. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 A resolution. Nature 2015, 517, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Popov, S.; Popova, E.; Inoue, M.; Weissenhorn, W.; Göttlinger, H.G. The ESCRT pathway and HIV-1 budding. Biochem. Soc. Trans. 2009, 37, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Schur, F.K.; Briggs, J.A. Retrovirus maturation-an extraordinary structural transformation. Curr. Opin. Virol. 2016, 18, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Eckwahl, M.J.; Arnion, H.; Kharytonchyk, S.; Zang, T.; Bieniasz, P.D.; Telesnitsky, A.; Wolin, S.L. Analysis of the human immunodeficiency virus-1 RNA packageome. RNA 2016, 22, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Chertova, E.; Chertov, O.; Coren, L.V.; Roser, J.D.; Trubey, C.M.; Bess, J.W., Jr.; Sowder, R.C., II; Barsov, E.; Hood, B.L.; Fisher, R.J.; et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 2006, 80, 9039–9052. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E. Cellular proteins detected in HIV-1. Rev. Med. Virol. 2008, 18, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.M.; Hunter, E. HIV transmission. Cold Spring Harb. Perspect. Med. 2012, 2, a006965. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Murooka, T.T.; Imle, A.; Mempel, T.R. Adding new dimensions: Towards an integrative understanding of HIV-1 spread. Nat. Rev. Microbiol. 2014, 12, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Koonin, E.V. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 2008, 23, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Ishizu, H.; Siomi, H.; Siomi, M.C. Biology of PIWI-interacting RNAs: New insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012, 26, 2361–2373. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Steinkraus, B.R.; Toegel, M.; Fulga, T.A. Tiny giants of gene regulation: Experimental strategies for microRNA functional studies. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 311–362. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. siRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Yi, R.; Cullen, B.R. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Doudna, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Tang, G. siRNA and miRNA: An insight into RISCs. Trends Biochem. Sci. 2005, 30, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Klattenhoff, C.; Theurkauf, W. Biogenesis and germline functions of piRNAs. Development 2008, 135, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.D.; Hannon, G.J. Small RNAs as guardians of the genome. Cell 2009, 136, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Londin, E.; Loher, P.; Telonis, A.G.; Quann, K.; Clark, P.; Jing, Y.; Hatzimichael, E.; Kirino, Y.; Honda, S.; Lally, M.; et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E1106–E1115. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Jo, M.H.; Choi, Y.G.; Park, J.; Kwon, S.C.; Hohng, S.; Kim, V.N.; Woo, J.S. Functional Anatomy of the Human Microprocessor. Cell 2015, 161, 1374–1387. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.C.; Nguyen, T.A.; Choi, Y.G.; Jo, M.H.; Hohng, S.; Kim, V.N.; Woo, J.S. Structure of Human DROSHA. Cell 2016, 164, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Auyeung, V.C.; Ulitsky, I.; McGeary, S.E.; Bartel, D.P. Beyond secondary structure: Primary-sequence determinants license pri-miRNA hairpins for processing. Cell 2013, 152, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Tambe, A.; Kidwell, M.A.; Noland, C.L.; Schneider, C.P.; Doudna, J.A. Dicer-TRBP complex formation ensures accurate mammalian microRNA biogenesis. Mol. Cell 2015, 57, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yeo, J.; Lee, J.H.; Cho, J.; Seo, D.; Kim, J.S.; Kim, V.N. Deletion of human tarbp2 reveals cellular microRNA targets and cell-cycle function of TRBP. Cell Rep. 2014, 9, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Macrae, I.J.; Zhou, K.; Li, F.; Repic, A.; Brooks, A.N.; Cande, W.Z.; Adams, P.D.; Doudna, J.A. Structural basis for double-stranded RNA processing by Dicer. Science 2006, 311, 195–198. [Google Scholar] [CrossRef] [PubMed]
- MacRae, I.J.; Zhou, K.; Doudna, J.A. Structural determinants of RNA recognition and cleavage by Dicer. Nat. Struct. Mol. Biol. 2007, 14, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Heo, I.; Tian, Y.; Simanshu, D.K.; Chang, H.; Jee, D.; Patel, D.J.; Kim, V.N. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature 2011, 475, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.W.; Guiley, K.Z.; De, N.; Potter, C.S.; Carragher, B.; MacRae, I.J. The molecular architecture of human Dicer. Nat. Struct. Mol. Biol. 2012, 19, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Hur, I.; Park, S.Y.; Kim, Y.K.; Suh, M.R.; Kim, V.N. The role of PACT in the RNA silencing pathway. EMBO J. 2006, 25, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Zhou, K.; Smith, A.M.; Noland, C.L.; Doudna, J.A. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013, 41, 6568–6576. [Google Scholar] [CrossRef] [PubMed]
- Czech, B.; Hannon, G.J. Small RNA sorting: Matchmaking for Argonautes. Nat. Rev. Genet. 2011, 12, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Kobayashi, M.; Yoda, M.; Sakaguchi, Y.; Katsuma, S.; Suzuki, T.; Tomari, Y. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol. Cell 2010, 39, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K. Anatomy of RISC: How do small RNAs and chaperones activate Argonaute proteins? Wiley Interdiscip. Rev. RNA 2016, 7, 637–660. [Google Scholar] [CrossRef] [PubMed]
- Kwak, P.B.; Tomari, Y. The N domain of Argonaute drives duplex unwinding during RISC assembly. Nat. Struct. Mol. Biol. 2012, 19, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Noland, C.L.; Doudna, J.A. Multiple sensors ensure guide strand selection in human RNAi pathways. RNA 2013, 19, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Rand, T.A.; Petersen, S.; Du, F.; Wang, X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 2005, 123, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Schirle, N.T.; Sheu-Gruttadauria, J.; MacRae, I.J. Structural basis for microRNA targeting. Science 2014, 346, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Chandradoss, S.D.; Schirle, N.T.; Szczepaniak, M.; MacRae, I.J.; Joo, C. A Dynamic Search Process Underlies MicroRNA Targeting. Cell 2015, 162, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Syed, A.P.; Bilen, B.; Zavolan, M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013, 23, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′UTR as in the 3′UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djuranovic, S.; Nahvi, A.; Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 2013, 340, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.H.; Ghoshal, K.; Villen, J.; Bartel, D.P. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.O.; Tomari, Y. The Functions of MicroRNAs: MRNA Decay and Translational Repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E.; Chung, W.J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian mirtron genes. Mol. Cell 2007, 28, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O'Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15163–15168. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D.; et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Veno, M.T.; Jensen, T.I.; Schaefer, A.; Damgaard, C.K.; Kjems, J. Argonaute-associated short introns are a novel class of gene regulators. Nat. Commun. 2016, 7, 11538. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, I.; Hansen, T.B. Biogenesis and Function of Ago-Associated RNAs. Trends Genet. 2017, 33, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Masaki, T.; Yamane, D.; McGivern, D.R.; Lemon, S.M. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2013, 110, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Arend, K.C.; Li, Y.; Yamane, D.; McGivern, D.R.; Kato, T.; Wakita, T.; Moorman, N.J.; Lemon, S.M. miR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 2015, 17, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Luna, J.M.; Liniger, M.; Nishiuchi, E.; Rozen-Gagnon, K.; Shlomai, A.; Auray, G.; Gerber, M.; Fak, J.; Keller, I.; et al. A Broad RNA Virus Survey Reveals Both miRNA Dependence and Functional Sequestration. Cell Host Microbe 2016, 19, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Scheel, T.K.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, L.; Yan, X.; Jin, W.; Wang, Y.; Chen, L.; Wu, E.; Ye, X.; Gao, G.F.; Wang, F.; et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1) -modulated P53 activity. Hepatology 2012, 55, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Wang, S.; Wu, B.; Hao, J.; Fan, H.; Ju, Y.; Ding, Y.; Chen, L.; Chu, X.; et al. Hepatitis B virus mRNA-mediated miR-122 inhibition upregulates PTTG1-binding protein, which promotes hepatocellular carcinoma tumor growth and cell invasion. J. Virol. 2013, 87, 2193–2205. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.C.; Yu, S.L.; Chen, J.J.; Chang, S.Y.; Yan, B.S.; Hong, Q.S.; Singh, S.; Kao, C.L.; Chen, H.Y.; Su, K.Y.; et al. Enterovirus-induced miR-141 contributes to shutoff of host protein translation by targeting the translation initiation factor eIF4E. Cell Host Microbe 2011, 9, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Linnstaedt, S.D.; Gottwein, E.; Skalsky, R.L.; Luftig, M.A.; Cullen, B.R. Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein-Barr virus. J. Virol. 2010, 84, 11670–11678. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Pan, Q.; Blencowe, B.J.; Claycomb, J.M.; Frappier, L. Epstein-Barr virus EBNA1 protein regulates viral latency through effects on let-7 microRNA and dicer. J. Virol. 2014, 88, 11166–11177. [Google Scholar] [CrossRef] [PubMed]
- Ellis-Connell, A.L.; Iempridee, T.; Xu, I.; Mertz, J.E. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. J. Virol. 2010, 84, 10329–10343. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Liu, H.; Rice, A.P. miR-132 enhances HIV-1 replication. Virology 2013, 438, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Nathans, R.; Chu, C.Y.; Serquina, A.K.; Lu, C.C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Sung, T.L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Sung, T.L.; Rice, A.P. Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Lioy, D.T.; Ma, L.; Impey, S.; Mandel, G.; Goodman, R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007, 10, 1513–1514. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.Y. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum. Mol. Genet. 2011, 20, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Chen, X.Y.; Wu, T.C.; Sang, W.W.; Ruan, Z. MiR-34a is involved in Tat-induced HIV-1 long terminal repeat (LTR) transactivation through the SIRT1/NFkappaB pathway. FEBS Lett. 2012, 586, 4203–4207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Wu, T.C.; Sang, W.W.; Ruan, Z. MiR-217 is involved in Tat-induced HIV-1 long terminal repeat (LTR) transactivation by down-regulation of SIRT1. Biochim. Biophys. Acta 2012, 1823, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, C.; Huang, J.; Cung, T.; Seiss, K.; Beamon, J.; Carrington, M.F.; Porter, L.C.; Burke, P.S.; Yang, Y.; et al. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J. Clin. Investig. 2011, 121, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.; Seharaseyon, J.; Dong, P.; Bour, S.; Marban, E. Mutual functional destruction of HIV-1 Vpu and host TASK-1 channel. Mol. Cell 2004, 14, 259–267. [Google Scholar] [CrossRef]
- Farberov, L.; Herzig, E.; Modai, S.; Isakov, O.; Hizi, A.; Shomron, N. MicroRNA-mediated regulation of p21 and TASK1 cellular restriction factors enhances HIV-1 infection. J. Cell Sci. 2015, 128, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.J.; Andrade, J.; Foxall, R.B.; Matoso, P.; Matos, A.M.; Soares, R.S.; Rocha, C.; Ramos, C.G.; Tendeiro, R.; Serra-Caetano, A.; et al. miRNA profiling of human naive CD4 T cells links miR-34c-5p to cell activation and HIV replication. EMBO J. 2017, 36, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.T. RNAi in the regulation of mammalian viral infections. BMC Biol. 2012, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Rana, T.M. RNA-based mechanisms regulating host-virus interactions. Immunol. Rev. 2013, 253, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R.; Cherry, S.; tenOever, B.R. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell Host Microbe 2013, 14, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Viruses and RNA interference: Issues and controversies. J. Virol. 2014, 88, 12934–12936. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, P.; Sklan, E.; Wilkins, C.; Burgon, T.; Samuel, M.A.; Lu, R.; Ansel, K.M.; Heissmeyer, V.; Einav, S.; Jackson, W.; et al. Six RNA viruses and forty-one hosts: Viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog. 2010, 6, e1000764. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Kincaid, R.P.; Arasappan, D.; Dowd, S.E.; Hunicke-Smith, S.P.; Sullivan, C.S. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA 2010, 16, 1540–1558. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Whisnant, A.W.; Kennedy, E.M.; Flores, O.; Cullen, B.R. Derivation and characterization of Dicer- and microRNA-deficient human cells. RNA 2014, 20, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Skalsky, R.L.; Kennedy, E.M.; Furuse, Y.; Whisnant, A.W.; Flores, O.; Schultz, K.L.; Putnam, N.; Barrows, N.J.; Sherry, B.; et al. Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. J. Virol. 2014, 88, 8065–8076. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Kincaid, R.P.; Phanaksri, T.; Burke, J.M.; Pare, J.M.; Cox, J.E.; Hsiang, T.Y.; Krug, R.M.; Sullivan, C.S. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 2013, 14, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Langlois, R.A.; Schmid, S.; Varble, A.; Shim, J.V.; Sachs, D.; tenOever, B.R. The Mammalian response to virus infection is independent of small RNA silencing. Cell Rep. 2014, 8, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; Ciaudo, C.; Marchais, A.; Li, Y.; Jay, F.; Ding, S.W.; Voinnet, O. Antiviral RNA interference in mammalian cells. Science 2013, 342, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, J.; Han, Y.; Fan, X.; Ding, S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science 2013, 342, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; Van der Veen, A.G.; Deddouche-Grass, S.; Rogers, N.C.; Merits, A.; Reis, E.S.C. Inactivation of the type I interferon pathway reveals long double-stranded RNA-mediated RNA interference in mammalian cells. EMBO J. 2016, 35, 2505–2518. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Basavappa, M.; Lu, J.; Dong, S.; Cronkite, D.A.; Prior, J.T.; Reinecker, H.C.; Hertzog, P.; Han, Y.; Li, W.X.; et al. Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat. Microbiol. 2016, 2, 16250. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, Y.; Zhang, Y.; Zhou, H.; Deng, Y.Q.; Li, X.F.; Miao, M.; Zhang, Q.; Zhong, B.; Hu, Y.; et al. Human Virus-Derived Small RNAs Can Confer Antiviral Immunity in Mammals. Immunity 2017, 46, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Tholen, L.E.; Overheul, G.J.; van Kuppeveld, F.J.M.; van Rij, R.P. Deletion of Cytoplasmic Double-Stranded RNA Sensors Does Not Uncover Viral Small Interfering RNA Production in Human Cells. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Lecellier, C.H.; Dunoyer, P.; Arar, K.; Lehmann-Che, J.; Eyquem, S.; Himber, C.; Saib, A.; Voinnet, O. A cellular microRNA mediates antiviral defense in human cells. Science 2005, 308, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Jing, Q.; Georgel, P.; New, L.; Chen, J.; Mols, J.; Kang, Y.J.; Jiang, Z.; Du, X.; Cook, R.; et al. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 2007, 27, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Ke, X.; Wang, M.; He, S.; Li, Q.; Zheng, C.; Zhang, Z.; Liu, Y.; Wang, H. Human microRNA hsa-miR-296-5p suppresses enterovirus 71 replication by targeting the viral genome. J. Virol. 2013, 87, 5645–5656. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.A.; Hsieh, W.Y.; Forster, T.; Blanc, M.; Lu, H.; Crick, P.J.; Yutuc, E.; Watterson, S.; Martin, K.; Griffiths, S.J.; et al. An Interferon Regulated MicroRNA Provides Broad Cell-Intrinsic Antiviral Immunity through Multihit Host-Directed Targeting of the Sterol Pathway. PLoS Biol. 2016, 14, e1002364. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Shapiro, J.S.; Sabin, L.R.; Pham, A.M.; Reyes, I.; Moss, B.; Cherry, S.; tenOever, B.R. Degradation of host microRNAs by poxvirus poly(A) polymerase reveals terminal RNA methylation as a protective antiviral mechanism. Cell Host Microbe 2012, 12, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, V.R.; Steel, L.F. A re-examination of global suppression of RNA interference by HIV-1. PLoS ONE 2011, 6, e17246. [Google Scholar] [CrossRef] [PubMed]
- Langlois, R.A.; Albrecht, R.A.; Kimble, B.; Sutton, T.; Shapiro, J.S.; Finch, C.; Angel, M.; Chua, M.A.; Gonzalez-Reiche, A.S.; Xu, K.; et al. MicroRNA-based strategy to mitigate the risk of gain-of-function influenza studies. Nat. Biotechnol. 2013, 31, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Heiss, B.L.; Maximova, O.A.; Pletnev, A.G. Insertion of microRNA targets into the flavivirus genome alters its highly neurovirulent phenotype. J. Virol. 2011, 85, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.T.; Nicot, C. miR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J. Biol. Chem. 2015, 290, 5381–5390. [Google Scholar] [CrossRef] [PubMed]
- Presloid, J.B.; Novella, I.S. RNA Viruses and RNAi: Quasispecies Implications for Viral Escape. Viruses 2015, 7, 3226–3240. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, M.; Scaria, V.; Pillai, B.; Brahmachari, S.K. Targets for human encoded microRNAs in HIV genes. Biochem. Biophys. Res. Commun. 2005, 337, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, J.K.; Khan, S.Z.; Soni, K.; Rawat, P.; Gupta, A.; Hariharan, M.; Scaria, V.; Lalwani, M.; Pillai, B.; Mitra, D.; et al. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology 2008, 5, 117. [Google Scholar] [CrossRef] [PubMed]
- Houzet, L.; Klase, Z.; Yeung, M.L.; Wu, A.; Le, S.Y.; Quinones, M.; Jeang, K.T. The extent of sequence complementarity correlates with the potency of cellular miRNA-mediated restriction of HIV-1. Nucleic Acids Res. 2012, 40, 11684–11696. [Google Scholar] [CrossRef] [PubMed]
- Whisnant, A.W.; Bogerd, H.P.; Flores, O.; Ho, P.; Powers, J.G.; Sharova, N.; Stevenson, M.; Chen, C.H.; Cullen, B.R. In-depth analysis of the interaction of HIV-1 with cellular microRNA biogenesis and effector mechanisms. MBio 2013, 4, e000193. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ye, L.; Hou, W.; Zhou, Y.; Wang, Y.J.; Metzger, D.S.; Ho, W.Z. Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 2009, 113, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Qu, X.; Zhou, X.; Shen, Y.; Ji, H.; Fu, Z.; Deng, J.; Lu, P.; Yu, W.; Lu, H.; et al. Two cellular microRNAs, miR-196b and miR-1290, contribute to HIV-1 latency. Virology 2015, 486, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Li, H.; Wu, X.; Covarrubias, M.; Scherer, L.; Meinking, K.; Luk, B.; Chomchan, P.; Alluin, J.; Gombart, A.F.; et al. Interplay between HIV-1 infection and host microRNAs. Nucleic Acids Res. 2012, 40, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Bennasser, Y.; Myers, T.G.; Jiang, G.; Benkirane, M.; Jeang, K.T. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology 2005, 2, 81. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Ho, D.D. HIV-1 dynamics in vivo: Implications for therapy. Nat. Rev. Microbiol. 2003, 1, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Mantri, C.K.; Pandhare Dash, J.; Mantri, J.V.; Dash, C.C. Cocaine enhances HIV-1 replication in CD4+ T cells by down-regulating MiR-125b. PLoS ONE 2012, 7, e51387. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.J.; Jia, Y.H.; Tian, R.R.; Ding, M.; Zhang, C.; Wang, J.H. Translation of Pur-alpha is targeted by cellular miRNAs to modulate the differentiation-dependent susceptibility of monocytes to HIV-1 infection. FASEB J. 2012, 26, 4755–4764. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Handrick, R.; Aschrafi, A.; Otte, K. Unveiling the principle of microRNA-mediated redundancy in cellular pathway regulation. RNA Biol. 2015, 12, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Shen, C.J.; Cohen, E.A.; Xiong, S.D.; Wang, J.H. miRNA-1236 inhibits HIV-1 infection of monocytes by repressing translation of cellular factor VprBP. PLoS ONE 2014, 9, e99535. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Rossi, F.; Sierra, L.J.; Gupta, A.; Navas-Martin, S.; Martin-Garcia, J. A role for microRNA-155 modulation in the anti-HIV-1 effects of Toll-like receptor 3 stimulation in macrophages. PLoS Pathog. 2012, 8, e1002937. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Chan, J.K.; Oh, E.; Heidersbach, A.J.; Hebbeler, A.M.; Chavez, L.; Verdin, E.; Rape, M.; Greene, W.C. MicroRNA-155 Reinforces HIV Latency. J. Biol. Chem. 2015, 290, 13736–13748. [Google Scholar] [CrossRef] [PubMed]
- Lodge, R.; Ferreira Barbosa, J.A.; Lombard-Vadnais, F.; Gilmore, J.C.; Deshiere, A.; Gosselin, A.; Wiche Salinas, T.R.; Bego, M.G.; Power, C.; Routy, J.P.; et al. Host MicroRNAs-221 and -222 Inhibit HIV-1 Entry in Macrophages by Targeting the CD4 Viral Receptor. Cell Rep. 2017, 21, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Pilakka-Kanthikeel, S.; Raymond, A.; Atluri, V.S.; Sagar, V.; Saxena, S.K.; Diaz, P.; Chevelon, S.; Concepcion, M.; Nair, M. Sterile alpha motif and histidine/aspartic acid domain-containing protein 1 (SAMHD1)-facilitated HIV restriction in astrocytes is regulated by miRNA-181a. J. Neuroinflamm. 2015, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Peng, X.; Liu, F.; Cheng, L.; Lu, X.; Yao, H.; Wu, H.; Wu, N. MicroRNA-181 expression regulates specific post-transcriptional level of SAMHD1 expression in vitro. Biochem. Biophys. Res. Commun. 2014, 452, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buchanan, E.L.; Myers, S.L.; McAlexander, M.A. miRNAs and SAMHD1 regulation in vitro and in a model of HIV CNS disease. J. Neuroinflamm. 2015, 12, 159. [Google Scholar] [CrossRef] [PubMed]
- Chable-Bessia, C.; Meziane, O.; Latreille, D.; Triboulet, R.; Zamborlini, A.; Wagschal, A.; Jacquet, J.M.; Reynes, J.; Levy, Y.; Saib, A.; et al. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 2009, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Adoro, S.; Cubillos-Ruiz, J.R.; Chen, X.; Deruaz, M.; Vrbanac, V.D.; Song, M.; Park, S.; Murooka, T.T.; Dudek, T.E.; Luster, A.D.; et al. IL-21 induces antiviral microRNA-29 in CD4 T cells to limit HIV-1 infection. Nat. Commun. 2015, 6, 7562. [Google Scholar] [CrossRef] [PubMed]
- MacKay, C.R.; Wang, J.P.; Kurt-Jones, E.A. Dicer’s role as an antiviral: Still an enigma. Curr. Opin. Immunol. 2014, 26, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Eckenfelder, A.; Segeral, E.; Pinzon, N.; Ulveling, D.; Amadori, C.; Charpentier, M.; Nidelet, S.; Concordet, J.P.; Zagury, J.F.; Paillart, J.C.; et al. Argonaute proteins regulate HIV-1 multiply spliced RNA and viral production in a Dicer independent manner. Nucleic Acids Res. 2017, 45, 4158–4173. [Google Scholar] [CrossRef] [PubMed]
- Bouttier, M.; Saumet, A.; Peter, M.; Courgnaud, V.; Schmidt, U.; Cazevieille, C.; Bertrand, E.; Lecellier, C.H. Retroviral GAG proteins recruit AGO2 on viral RNAs without affecting RNA accumulation and translation. Nucleic Acids Res. 2012, 40, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Vongrad, V.; Imig, J.; Mohammadi, P.; Kishore, S.; Jaskiewicz, L.; Hall, J.; Gunthard, H.F.; Beerenwinkel, N.; Metzner, K.J. HIV-1 RNAs are Not Part of the Argonaute 2 Associated RNA Interference Pathway in Macrophages. PLoS ONE 2015, 10, e0132127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.K.; Sengupta, P.; Waki, K.; Van Engelenburg, S.B.; Ochiya, T.; Ablan, S.D.; Freed, E.O.; Lippincott-Schwartz, J. MicroRNA binding to the HIV-1 Gag protein inhibits Gag assembly and virus production. Proc. Natl. Acad. Sci. USA 2014, 111, E2676–E2683. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cullen, B.R. Analysis of the interaction of primate retroviruses with the human RNA interference machinery. J. Virol. 2007, 81, 12218–12226. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Das, A.T.; Berkhout, B. Retroviral microRNAs. Curr. Opin. Virol. 2014, 7, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Viruses and microRNAs. Nat. Genet. 2006, 38, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Cullen, B.R. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 2008, 3, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Sullivan, C.S. Virus-encoded microRNAs: An overview and a look to the future. PLoS Pathog. 2012, 8, e1003018. [Google Scholar] [CrossRef] [PubMed]
- Rosewick, N.; Momont, M.; Durkin, K.; Takeda, H.; Caiment, F.; Cleuter, Y.; Vernin, C.; Mortreux, F.; Wattel, E.; Burny, A.; et al. Deep sequencing reveals abundant noncanonical retroviral microRNAs in B-cell leukemia/lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2306–2311. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Chen, Y.; Cox, J.E.; Rethwilm, A.; Sullivan, C.S. Noncanonical microRNA (miRNA) biogenesis gives rise to retroviral mimics of lymphoproliferative and immunosuppressive host miRNAs. MBio 2014, 5, e00074. [Google Scholar] [CrossRef] [PubMed]
- Whisnant, A.W.; Kehl, T.; Bao, Q.; Materniak, M.; Kuzmak, J.; Lochelt, M.; Cullen, B.R. Identification of novel, highly expressed retroviral microRNAs in cells infected by bovine foamy virus. J. Virol. 2014, 88, 4679–4686. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Hamaidia, M.; de Brogniez, A.; Gutierrez, G.; Renotte, N.; Reichert, M.; Trono, K.; Willems, L. Bovine Leukemia Virus Small Noncoding RNAs Are Functional Elements That Regulate Replication and Contribute to Oncogenesis In Vivo. PLoS Pathog. 2016, 12, e1005588. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Radke, K.; Sigala, T.J.; Grossman, D. Transcription of bovine leukemia virus in peripheral blood cells obtained during early infection in vivo. Microb. Pathog. 1992, 12, 319–331. [Google Scholar] [CrossRef]
- Rovnak, J.; Casey, J.W. Assessment of bovine leukemia virus transcripts in vivo. J. Virol. 1999, 73, 8890–8897. [Google Scholar] [PubMed]
- Yao, Y.; Smith, L.P.; Nair, V.; Watson, M. An avian retrovirus uses canonical expression and processing mechanisms to generate viral microRNA. J. Virol. 2014, 88, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Bennasser, Y.; Le, S.Y.; Yeung, M.L.; Jeang, K.T. HIV-1 encoded candidate micro-RNAs and their cellular targets. Retrovirology 2004, 1, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couturier, J.P.; Root-Bernstein, R.S. HIV may produce inhibitory microRNAs (miRNAs) that block production of CD28, CD4 and some interleukins. J. Theor. Biol. 2005, 235, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Ito, M.; Tsutsumi, Y.; Ichikawa, Y.; Okuyama, H.; Brisibe, E.A.; Saksena, N.K.; Fujii, Y.R. HIV-1 nef suppression by virally encoded microRNA. Retrovirology 2004, 1, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omoto, S.; Fujii, Y.R. Regulation of human immunodeficiency virus 1 transcription by nef microRNA. J. Gen. Virol. 2005, 86, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D.; Ahlawat, A.; Gupta, S.D. HIV-1 genome-encoded hiv1-mir-H1 impairs cellular responses to infection. Mol. Cell. Biochem. 2009, 323, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Lamers, S.L.; Fogel, G.B.; McGrath, M.S. HIV-miR-H1 evolvability during HIV pathogenesis. Biosystems 2010, 101, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Klase, Z.; Winograd, R.; Davis, J.; Carpio, L.; Hildreth, R.; Heydarian, M.; Fu, S.; McCaffrey, T.; Meiri, E.; Ayash-Rashkovsky, M.; et al. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology 2009, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Plante, I.; Landry, P.; Barat, C.; Janelle, M.E.; Flamand, L.; Tremblay, M.J.; Provost, P. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008, 36, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Vigneault-Edwards, J.; Letourneau, K.; Gobeil, L.A.; Plante, I.; Burnett, J.C.; Rossi, J.J.; Provost, P. Regulation of host gene expression by HIV-1 TAR microRNAs. Retrovirology 2013, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, M.; Geng, G.; Liu, B.; Huang, Z.; Luo, H.; Zhou, J.; Guo, X.; Cai, W.; Zhang, H. A novel HIV-1-encoded microRNA enhances its viral replication by targeting the TATA box region. Retrovirology 2014, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Schopman, N.C.; Willemsen, M.; Liu, Y.P.; Bradley, T.; van Kampen, A.; Baas, F.; Berkhout, B.; Haasnoot, J. Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012, 40, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Cullen, B.R. How do viruses avoid inhibition by endogenous cellular microRNAs? PLoS Pathog. 2013, 9, e1003694. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Mullokandov, G.; Baccarini, A.; Ruzo, A.; Jayaprakash, A.D.; Tung, N.; Israelow, B.; Evans, M.J.; Sachidanandam, R.; Brown, B.D. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat. Methods 2012, 9, 840–846. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.J.; Ribeiro, R.M.; Perelson, A.S. Current estimates for HIV-1 production imply rapid viral clearance in lymphoid tissues. PLoS Comput. Biol. 2010, 6, e1000906. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Kennedy, E.M.; Whisnant, A.W.; Cullen, B.R. Induced Packaging of Cellular MicroRNAs into HIV-1 Virions Can Inhibit Infectivity. MBio 2017, 8, e02125-16. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target interactions—Beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Ruegger, S.; Grosshans, H. MicroRNA turnover: When, how, and why. Trends Biochem. Sci. 2012, 37, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Aguado, L.C.; Schmid, S.; Sachs, D.; Shim, J.V.; Lim, J.K.; tenOever, B.R. microRNA Function Is Limited to Cytokine Control in the Acute Response to Virus Infection. Cell Host Microbe 2015, 18, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Broderick, J.A.; Zamore, P.D. Competitive endogenous RNAs cannot alter microRNA function in vivo. Mol. Cell 2014, 54, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Dinger, M.E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 2016, 17, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Cazalla, D.; Yario, T.; Steitz, J.A. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, J.; Jiao, T.; Li, Z.; Peng, J.; Cui, Z.; Ye, X. Hepatitis B virus inhibits apoptosis of hepatoma cells by sponging the MicroRNA 15a/16 cluster. J. Virol. 2013, 87, 13370–13378. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Helwak, A.; Miesen, P.; Santhakumar, D.; Borger, J.G.; Kudla, G.; Grey, F.; Tollervey, D.; Buck, A.H. Murine cytomegalovirus encodes a miR-27 inhibitor disguised as a target. Proc. Natl. Acad. Sci. USA 2012, 109, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Marcinowski, L.; Tanguy, M.; Krmpotic, A.; Radle, B.; Lisnic, V.J.; Tuddenham, L.; Chane-Woon-Ming, B.; Ruzsics, Z.; Erhard, F.; Benkartek, C.; et al. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog. 2012, 8, e1002510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Song, J.; Kim, S.; Kim, J.; Hong, Y.; Kim, Y.; Kim, D.; Baek, D.; Ahn, K. Selective degradation of host MicroRNAs by an intergenic HCMV noncoding RNA accelerates virus production. Cell Host Microbe 2013, 13, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.S.; Dohjima, T.; Bauer, G.; Li, H.; Li, M.J.; Ehsani, A.; Salvaterra, P.; Rossi, J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 2002, 20, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Novina, C.D.; Murray, M.F.; Dykxhoorn, D.M.; Beresford, P.J.; Riess, J.; Lee, S.K.; Collman, R.G.; Lieberman, J.; Shankar, P.; Sharp, P.A. siRNA-directed inhibition of HIV-1 infection. Nat. Med. 2002, 8, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Jacque, J.M.; Triques, K.; Stevenson, M. Modulation of HIV-1 replication by RNA interference. Nature 2002, 418, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Capodici, J.; Kariko, K.; Weissman, D. Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J. Immunol. 2002, 169, 5196–5201. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Shank, P.R.; Ramratnam, B. Promoter choice affects the potency of HIV-1 specific RNA interference. Nucleic Acids Res. 2003, 31, 5033–5038. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Pusch, O.; Boden, D.; Silbermann, R.; Lee, F.; Tucker, L.; Ramratnam, B. Nucleotide sequence homology requirements of HIV-1-specific short hairpin RNA. Nucleic Acids Res. 2003, 31, 6444–6449. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Westerhout, E.M.; Ooms, M.; Vink, M.; Das, A.T.; Berkhout, B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005, 33, 796–804. [Google Scholar] [CrossRef] [PubMed]
- ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Westerhout, E.M.; Berkhout, B. A systematic analysis of the effect of target RNA structure on RNA interference. Nucleic Acids Res. 2007, 35, 4322–4330. [Google Scholar] [CrossRef] [PubMed]
- Nevot, M.; Martrus, G.; Clotet, B.; Martinez, M.A. RNA interference as a tool for exploring HIV-1 robustness. J. Mol. Biol. 2011, 413, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.S.; Pham, N.P.; Schaffer, D.V. HIV develops indirect cross-resistance to combinatorial RNAi targeting two distinct and spatially distant sites. Mol. Ther. 2012, 20, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liang, Z.; Kong, X. Efficacy Analysis of Combinatorial siRNAs against HIV Derived from One Double Hairpin RNA Precursor. Front. Microbiol. 2017, 8, 1651. [Google Scholar] [CrossRef] [PubMed]
- Didiano, D.; Hobert, O. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat. Struct. Mol. Biol. 2006, 13, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N. Re-thinking miRNA-mRNA interactions: Intertwining issues confound target discovery. Bioessays 2015, 37, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Lee, R.; Williams, P.; Chan, C.Y.; Ambros, V.; Ding, Y. Potent effect of target structure on microRNA function. Nat. Struct. Mol. Biol. 2007, 14, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Marin, R.M.; Vanicek, J. Efficient use of accessibility in microRNA target prediction. Nucleic Acids Res. 2011, 39, 19–29. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNA | Sequence (5′-3′) | Target Region on HIV-1 RNA | Targeted Sequence on HIV-1 RNA (5′-3′) | Effect on HIV-1 Replication | Reference | |
|---|---|---|---|---|---|---|
| 1 | miR-149-5p | UCUGGCUCCGUGUCUUCACUCCC | Gag | GGGAGTGGGGGGACCCGGCCATA | Negative | Houzet et al. 2012 [221] |
| 2 | miR-423-3p | AGCUCGGUCUGAGGCCCCUCAGU | Gag | UAUAAAACUCUAAGAGCCGAGCA | Negative | Whisnant et al. 2013 [222] |
| 3 | miR-92a-3p | UAUUGCACUUGUCCCGGCCUGU | Pol | GCATTGGGAATCATTCAAGCACAACCA | Negative | Houzet et al. 2012 [221] |
| 4 | miR-155-5p | UUAAUGCUAAUCGUGAUAGGGGU | Vif | GUACUUGGCACUAGCAGCAUUAA | Negative | Whisnant et al. 2013 [222] |
| 5 | miR-133b | UUUGGUCCCCUUCAACCAGCUA | Env | GACCTGGATGGAGTGGGACAGA | Negative | Houzet et al. 2012 [221] |
| 6 | miR-138-5p | AGCUGGUGUUGUGAAUCAGGCCG | Env | ATTGAAGAATCGCAAAACCAGCA | Negative | Houzet et al. 2012 [221] |
| 7 | miR-125b-5p | UCCCUGAGACCCUAACUUGUGA | Env | ACGAGGAUUGUGGAACUUCUGGGACGCAGGGG | Negative | Huang et al. 2007 [179] |
| 8 | miR-150-5p | UCUCCCAACCCUUGUACCAGUG | Env | UCUGGGACGCAGGGGGUGGGAA | Negative | Huang et al. 2007 [179]; Wang et al. 2009 [223] |
| 9 | miR-1290 | UGGAUUUUUGGAUCAGGGA | Nef/3′UTR | UCCUUGAUCTGUGGAUCUA | Negative | Wang et al. 2015 [224] |
| 10 | miR-196b | UAGGUAGUUUCCUGUUGUUGGG | Nef/3′UTR | CUACCACACACAAGGCUACUUC | Negative | Wang et al. 2015 [224] |
| 11 | miR-223-3p | UGUCAGUUUGUCAAAUACCCCA | Nef/3′UTR | AGGGGUCAGAUAUCCACUGACC | Negative | Huang et al. 2007 [179]; Sun et al. 2012 [225]; Wang et al. 2009 [223] |
| 12 | miR-29b-3p | UAGCACCAUUUGAAAUCAGUGUU | Nef/3′UTR | AUACCCACUGACCUUUGGAUGGUGCUU | Negative | Sun et al. 2012 [225] |
| 13 | miR-29a-3p | UAGCACCAUCUGAAAUCGGUUA | Nef/3′UTR | CACUGACCUUUGGAUGGUGCUU | Negative | Ahluwalia et al. 2008 [220]; Nathans et al. 2009 [180] |
| 14 | miR-326 | CCUCUGGGCCCUUCCUCCAG | Nef/3′UTR | TGGGATGGAGGACCCGGAGG | Negative | Houzet et al. 2012 [221] |
| 15 | miR-382-5p | GAAGUUGUUCGUGGUGGAUUCG | U3/3′UTR | CGAGCUUGCUACAAGGGACUUU | Negative | Huang et al. 2007 [179]; Wang et al. 2009 [223] |
| 16 | miR-28-5p | AAGGAGCUCACAGUCUAUUGAG | U3/3′UTR | AUCUGAGCCUGGGAGCUCUC | Negative | Huang et al. 2007 [179]; Wang et al. 2009 [223] |
| miRNA | Target HDF/Cellular mRNA | Effect on HIV-1 Replication | Reference | |
|---|---|---|---|---|
| 1 | miR-17-5p | PCAF 3′UTR | Reduction in HIV-1 infection in PBMCs and Jurkat cells | Triboulet et al. 2007 [181] |
| 2 | miR-20a | PCAF 3′UTR | Reduction in HIV-1 infection in PBMCs and Jurkat cells | Triboulet et al. 2007 [181] |
| 3 | miR-198 | Cyclin T1 3′UTR | Impaired HIV-1 replication in monocytes | Sung and Rice, 2009 [182] |
| 4 | miR-15a | Pur-alpha 3′UTR | Impaired HIV-1 replication in monocytes | Shen et al. 2012 [229] |
| 5 | miR-15b | Pur-alpha 3′UTR | Impaired HIV-1 replication in monocytes | Shen et al. 2012 [229] |
| 6 | miR-16 | Pur-alpha 3′UTR | Impaired HIV-1 replication in monocytes | Shen et al. 2012 [229] |
| 7 | miR-20a | Pur-alpha 3′UTR | Impaired HIV-1 replication in monocytes | Shen et al. 2012 [229] |
| 8 | miR-93 | Pur-alpha 3′UTR | Impaired HIV-1 replication in monocytes | Shen et al. 2012 [229] |
| 9 | miR-106b | Pur-alpha 3′UTR | Impaired HIV-1 replication in monocytes | Shen et al. 2012 [229] |
| 10 | miR-155 | ADAM10 3′UTR, NUP153 3′UTR, LEDGF/p75 3′UTRTRIM32 3′UTR SAMHD1 | Reduction in HIV-1 late RT products and viral DNA integration in MDMs Promotes reactivation of latent HIV-1 via NF-kB signaling in J-Lat 5A8 cells Overexpression of miR-155 enhanced HIV-1 replication in astrocytes | Swaminathan et al. 2012 [232] Ruelas et al. 2015 [233] Pilakka-Kanthikeel et al. 2015 [235] |
| 11 | miR-27b | Cyclin T1 3′UTR | Impaired HIV-1 replication in resting CD4+ T-cells | Chiang et al. 2012 [183] |
| 12 | miR-29b | Cyclin T1 3′UTR | Impaired HIV-1 replication in resting CD4+ T-cells | Chiang et al. 2012 [183] |
| 13 | miR-150 | Cyclin T1 3′UTR | Impaired HIV-1 replication in resting CD4+ T-cells | Chiang et al. 2012 [183] |
| 14 | miR-223 | Cyclin T1 3′UTR | Impaired HIV-1 replication in resting CD4+ T-cells | Chiang et al. 2012 [183] |
| 15 | miR-1236 | VprBP 3′UTR | Impaired HIV-1 replication in monocytes | Ma et al. 2014 [231] |
| 16 | miR-181a | SAMHD1 | Overexpression of miR-181a enhanced HIV-1 replication in astrocytes | Pilakka-Kanthikeel et al. 2015 [235] |
| 17 | miR-132 | MeCP2 | Increased HIV-1 infection in Jurkat T-cells | Chiang et al. 2013 [178] |
| 18 | Let-7c | P21 3′UTR | Increased HIV-1 replication in JLTRG-R5 and HeLa-CCR5 cells | Farberov et al. 2015 [190] |
| 19 | miR-124a | TASK1 3′UTR | Increased HIV-1 replication in JLTRG-R5 and HeLa-CCR5 cells | Farberov et al. 2015 [190] |
| 20 | miR-34a-5p | TASK1 3′UTR | Increased HIV-1 replication in JLTRG-R5 and HeLa-CCR5 cells | Farberov et al. 2015 [190] |
| 21 | miR-34c-5p | Several genes involved in TCR signaling and activation of naïve CD4+ T-cells | Increased HIV-1 replication in Jurkat T-cells | Amaral et al. 2017 [191] |
| 22 | miR-221 | CD4 3′UTR | Inhibition of HIV-1 entry in macrophages | Lodge et al. 2017 [234] |
| 23 | miR-222 | CD4 3′UTR | Inhibition of HIV-1 entry in macrophages | Lodge et al. 2017 [234] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balasubramaniam, M.; Pandhare, J.; Dash, C. Are microRNAs Important Players in HIV-1 Infection? An Update. Viruses 2018, 10, 110. https://doi.org/10.3390/v10030110
Balasubramaniam M, Pandhare J, Dash C. Are microRNAs Important Players in HIV-1 Infection? An Update. Viruses. 2018; 10(3):110. https://doi.org/10.3390/v10030110
Chicago/Turabian StyleBalasubramaniam, Muthukumar, Jui Pandhare, and Chandravanu Dash. 2018. "Are microRNAs Important Players in HIV-1 Infection? An Update" Viruses 10, no. 3: 110. https://doi.org/10.3390/v10030110
APA StyleBalasubramaniam, M., Pandhare, J., & Dash, C. (2018). Are microRNAs Important Players in HIV-1 Infection? An Update. Viruses, 10(3), 110. https://doi.org/10.3390/v10030110