Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Culture
2.2. Virus Production and Infections
2.3. Extracellular Flux Measurements
2.4. ELISA and Western Blotting
2.5. Flow Cytometry
2.6. qPCR
2.7. 1H-NMR Analysis
2.8. Glycolytic Enzyme Kinetic Assay
2.9. Statistical Analyses
2.10. Ethical Approval
3. Results
3.1. Activation-Dependent Expression of Multiple GLUT Proteins in Human CD4+ T Cells
3.2. Upregulation of GLUT Proteins in HIV-1 Infected CD4+ T Cells
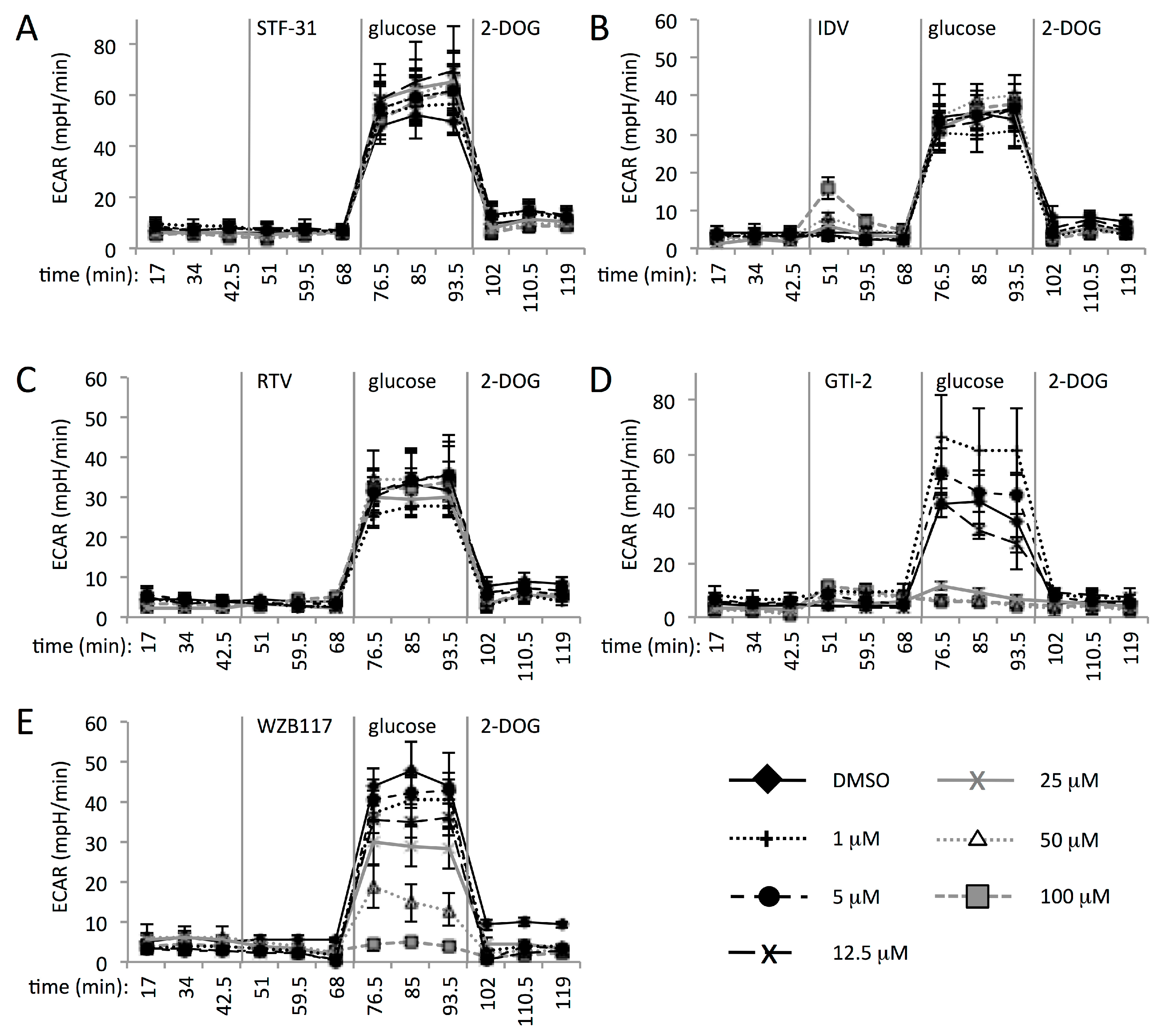
3.3. Multiple GLUT Proteins Mediate Glucose Transport into Human CD4+ T Cells
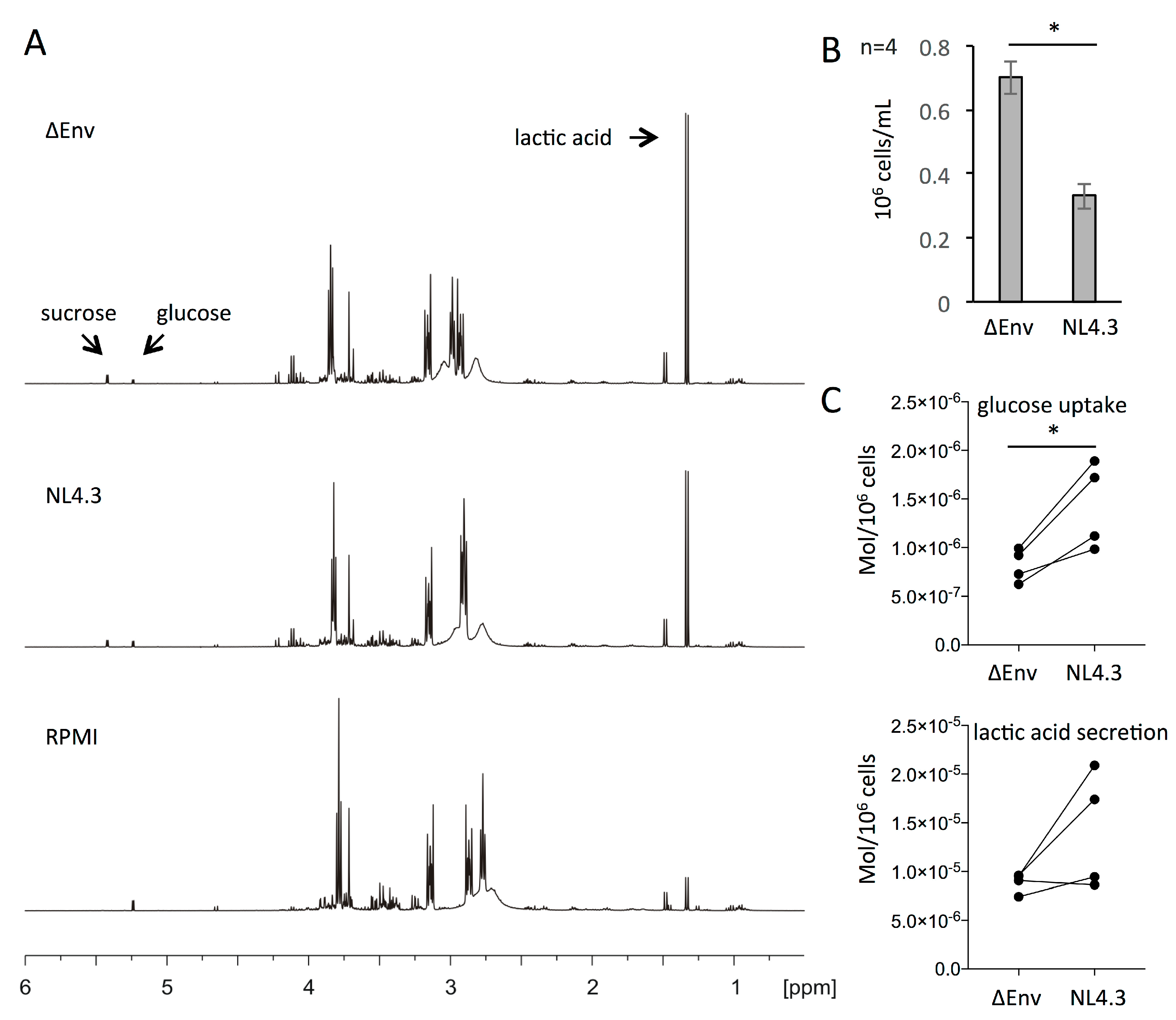
3.4. Increased Glucose Consumption of HIV-1 Infected CD4+ T Cells
3.5. HIV-1 Infected Cells Have Increased Hexokinase Activity That Is Mediated by Upregulation of HK1
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDougal, J.S.; Mawle, A.; Cort, S.P.; Nicholson, J.K.; Cross, G.D.; Scheppler-Campbell, J.A.; Hicks, D.; Sligh, J. Cellular tropism of the human retrovirus HTLV-III/LAV. I. Role of T cell activation and expression of the T4 antigen. J. Immunol. 1985, 135, 3151–3162. [Google Scholar] [PubMed]
- Zagury, D.; Bernard, J.; Leonard, R.; Cheynier, R.; Feldman, M.; Sarin, P.S.; Gallo, R.C. Long-term cultures of HTLV-III-infected T cells: A model of cytopathology of T-cell depletion in AIDS. Science 1986, 231, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Gowda, S.D.; Stein, B.S.; Mohagheghpour, N.; Benike, C.J.; Engleman, E.G. Evidence that T cell activation is required for HIV-1 entry in CD4+ lymphocytes. J. Immunol. 1989, 142, 773–780. [Google Scholar] [PubMed]
- Korin, Y.D.; Zack, J.A. Nonproductive human immunodeficiency virus type 1 infection in nucleoside-treated G0 lymphocytes. J. Virol. 1999, 73, 6526–6532. [Google Scholar] [PubMed]
- Plesa, G.; Dai, J.; Baytop, C.; Riley, J.L.; June, C.H.; O’Doherty, U. Addition of deoxynucleosides enhances human immunodeficiency virus type 1 integration and 2LTR formation in resting CD4+ T cells. J. Virol. 2007, 81, 13938–13942. [Google Scholar] [CrossRef] [PubMed]
- Frauwirth, K.A.; Thompson, C.B. Regulation of T lymphocyte metabolism. J. Immunol. 2004, 172, 4661–4665. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K.N.; Powell, J.D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 2014, 14, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Maciolek, J.A.; Pasternak, J.A.; Wilson, H.L. Metabolism of activated T lymphocytes. Curr. Opin. Immunol. 2014, 27, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, A.; Kavanagh Williamson, M.; Huthoff, H. HIV-1 pathogenicity and virion production are dependent on the metabolic phenotype of activated CD4+ T cells. Retrovirology 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Vander Heiden, M.G.; Harris, M.H.; Frauwirth, K.A.; Thompson, C.B. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol. Cell 2000, 6, 683–692. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Maciver, N.J.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Coloff, J.L.; Rathmell, J.C. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 2008, 84, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Loisel-Meyer, S.; Swainson, L.; Craveiro, M.; Oburoglu, L.; Mongellaz, C.; Costa, C.; Martinez, M.; Cosset, F.L.; Battini, J.L.; Herzenberg, L.A.; et al. Glut1-mediated glucose transport regulates HIV infection. Proc. Natl. Acad. Sci. USA 2012, 109, 2549–2554. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Ostrowski, M.; Gouillou, M.; Tsai, L.; Yu, D.; Zhou, J.; Henstridge, D.C.; Maisa, A.; Hearps, A.C.; Lewin, S.R.; et al. Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. AIDS 2014, 28, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Anzinger, J.J.; Zhou, J.; Gouillou, M.; Landay, A.; Jaworowski, A.; McCune, J.M.; Crowe, S.M. Glucose transporter 1-expressing proinflammatory monocytes are elevated in combination antiretroviral therapy-treated and untreated HIV+ subjects. J. Immunol. 2014, 193, 5595–5603. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, T.R.; Hanna, D.B.; Kaplan, R.C.; Kizer, J.R.; Durkin, H.G.; Young, M.A.; Nowicki, M.J.; Tien, P.C.; Golub, E.T.; Floris-Moore, M.A.; et al. Increased glucose transporter-1 expression on intermediate monocytes from HIV-infected women with subclinical cardiovascular disease. AIDS 2017, 31, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Uldry, M.; Thorens, B. The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch. 2004, 447, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life 2010, 62, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Swainson, L.; Kinet, S.; Manel, N.; Battini, J.L.; Sitbon, M.; Taylor, N. Glucose transporter 1 expression identifies a population of cycling CD4+ CD8+ human thymocytes with high CXCR4-induced chemotaxis. Proc. Natl. Acad. Sci. USA 2005, 102, 12867–12872. [Google Scholar] [CrossRef] [PubMed]
- Cretenet, G.; Clerc, I.; Matias, M.; Loisel, S.; Craveiro, M.; Oburoglu, L.; Kinet, S.; Mongellaz, C.; Dardalhon, V.; Taylor, N. Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci. Rep. 2016, 6, 24129. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Hubbard, B.; Shevach, E.M. Foxp3-mediated inhibition of Akt inhibits Glut1 (glucose transporter 1) expression in human T regulatory cells. J. Leukoc. Biol. 2015, 97, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Jung, C.Y.; Lee, T.P.; Liu, H.; Mookerjee, B.K. Changes in glucose transport and transporter isoforms during the activation of human peripheral blood lymphocytes by phytohemagglutinin. J. Immunol. 1994, 152, 2660–2668. [Google Scholar] [PubMed]
- Estrada, D.E.; Elliott, E.; Zinman, B.; Poon, I.; Liu, Z.; Klip, A.; Daneman, D. Regulation of glucose transport and expression of Glut3 transporters in human circulating mononuclear cells: Studies in cells from insulin-dependent diabetic and nondiabetic individuals. Metabolism 1994, 43, 591–598. [Google Scholar] [CrossRef]
- Fu, Y.; Maianu, L.; Melbert, B.R.; Garvey, W.T. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: A role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol. Dis. 2004, 32, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Piatkiewicz, P.; Czech, A.; Taton, J.; Gorski, A. Investigations of cellular glucose transport and its regulation under the influence of insulin in human peripheral blood lymphocytes. Endokrynol. Pol. 2010, 61, 182–187. [Google Scholar] [PubMed]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Marko, A.J.; Miller, R.A.; Kelman, A.; Frauwirth, K.A. Induction of glucose metabolism in stimulated T lymphocytes is regulated by mitogen-activated protein kinase signaling. PLoS ONE 2010, 5, e15425. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Rathmell, J.C.; Macintyre, A.N. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS ONE 2014, 9, e104104. [Google Scholar] [CrossRef] [PubMed]
- Dziurla, R.; Gaber, T.; Fangradt, M.; Hahne, M.; Tripmacher, R.; Kolar, P.; Spies, C.M.; Burmester, G.R.; Buttgereit, F. Effects of hypoxia and/or lack of glucose on cellular energy metabolism and cytokine production in stimulated human CD4+ T lymphocytes. Immunol. Lett. 2010, 131, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Sounidaki, M.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. In human alloreactive CD4(+) T-cells, dichloroacetate inhibits aerobic glycolysis, induces apoptosis and favors differentiation towards the regulatory T-cell subset instead of effector T-cell subsets. Mol. Med. Rep. 2016, 13, 3370–3376. [Google Scholar] [CrossRef] [PubMed]
- Hollenbaugh, J.A.; Munger, J.; Kim, B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC-MS/MS analysis. Virology 2011, 415, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hollenbaugh, J.A.; Montero, C.; Schinazi, R.F.; Munger, J.; Kim, B. Metabolic profiling during HIV-1 and HIV-2 infection of primary human monocyte-derived macrophages. Virology 2016, 491, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Vollers, S.S.; Carruthers, A. Sequence determinants of Glut1-mediated accelerated-exchange transport: Analysis by homology-scanning mutagenesis. J. Biol. Chem. 2012, 287, 42533–42544. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh Williamson, M.; Huthoff, H. Various qPCR, Western Blotting, Flow Cytometry and Cell Culture Data as Described in the Text But Not of Sufficient Interest to be Included as Full or Supplementary Figures; King’s College London: London, UK, 2018. [Google Scholar]
- James, D.E.; Brown, R.; Navarro, J.; Pilch, P.F. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 1988, 333, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.N.; Farmer, S.R.; Pilch, P.F. Glut4 storage vesicles without Glut4: Transcriptional regulation of insulin-dependent vesicular traffic. Mol. Cell. Biol. 2004, 24, 7151–7162. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Alizadeh, A.A.; Rando, O.J.; Liu, C.L.; Stankunas, K.; Botstein, D.; Crabtree, G.R.; Brown, P.O. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc. Natl. Acad. Sci. USA 2002, 99, 11796–11801. [Google Scholar] [CrossRef] [PubMed]
- Grolleau, A.; Bowman, J.; Pradet-Balade, B.; Puravs, E.; Hanash, S.; Garcia-Sanz, J.A.; Beretta, L. Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J. Biol. Chem. 2002, 277, 22175–22184. [Google Scholar] [CrossRef] [PubMed]
- Brodie, T.; Brenna, E.; Sallusto, F. OMIP-018: Chemokine receptor expression on human T helper cells. Cytom. A 2013, 83, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Heitmeier, M.; Hruz, P.W.; Shanmugam, M. Evaluating the efficacy of Glut inhibitors using a seahorse extracellular flux analyzer. Methods Mol. Biol. 2018, 1713, 69–75. [Google Scholar] [PubMed]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chu, P.C.; Yang, C.N.; Yan, R.; Chuang, Y.C.; Kulp, S.K.; Chen, C.S. Development of a novel class of glucose transporter inhibitors. J. Med. Chem. 2012, 55, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, W.; Cao, Y.; Liu, Y.; Bergmeier, S.; Chen, X. Small compound inhibitors of basal glucose transport inhibit cell proliferation and induce apoptosis in cancer cells via glucose-deprivation-like mechanisms. Cancer Lett. 2010, 298, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Chen, X.; Bergmeier, S.C. Novel inhibitors of basal glucose transport as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Hresko, R.C.; Hruz, P.W. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of gluts with differing affinities for Glut1 and Glut4. PLoS ONE 2011, 6, e25237. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Hruz, P.W.; Mueckler, M. Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. AIDS 2002, 16, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Hruz, P.W.; Mueckler, M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J. Biol. Chem. 2000, 275, 20251–20254. [Google Scholar] [CrossRef] [PubMed]
- Ojelabi, O.A.; Lloyd, K.P.; Simon, A.H.; De Zutter, J.K.; Carruthers, A. WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate) inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J. Biol. Chem. 2016, 291, 26762–26772. [Google Scholar] [CrossRef] [PubMed]
- Rudich, A.; Konrad, D.; Torok, D.; Ben-Romano, R.; Huang, C.; Niu, W.; Garg, R.R.; Wijesekara, N.; Germinario, R.J.; Bilan, P.J.; et al. Indinavir uncovers different contributions of Glut4 and Glut1 towards glucose uptake in muscle and fat cells and tissues. Diabetologia 2003, 46, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Kurapati, K.R.; Samikkannu, T.; Atluri, V.S.; Nair, M.P. Cell cycle checkpoints and pathogenesis of HIV-1 infection: A brief overview. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Young, C.D.; Lewis, A.S.; Rudolph, M.C.; Ruehle, M.D.; Jackman, M.R.; Yun, U.J.; Ilkun, O.; Pereira, R.; Abel, E.D.; Anderson, S.M. Modulation of glucose transporter 1 (Glut1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PLoS ONE 2011, 6, e23205. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Cheng, J.C.; Singhal, S.; Krett, N.L.; Rosen, S.T.; Shanmugam, M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: Implications for glucose transporter-directed therapy. Blood 2012, 119, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Westerterp, M.; Bhagwat, N.; Cremers, S.; Shih, A.; Abdel-Wahab, O.; Lutjohann, D.; Randolph, G.J.; Levine, R.L.; Tall, A.R.; et al. HDL and Glut1 inhibition reverse a hypermetabolic state in mouse models of myeloproliferative disorders. J. Exp. Med. 2013, 210, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Yang, K.; Zhang, M.; Zou, M.; Bai, E.; Ma, Q.; Xu, R. Specific protein 1 depletion attenuates glucose uptake and proliferation of human glioma cells by regulating GLUT3 expression. Oncol. Lett. 2016, 12, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.M.; Malim, M.H. Snapshot: HIV-1 proteins. Cell 2008, 133, 742. [Google Scholar] [CrossRef] [PubMed]
- De Giorgis, V.; Varesio, C.; Baldassari, C.; Piazza, E.; Olivotto, S.; Macasaet, J.; Balottin, U.; Veggiotti, P. Atypical manifestations in Glut1 deficiency syndrome. J. Child Neurol. 2016, 31, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, L.R.; Maldarelli, F.; Chamoun, G.; Schilling, B.; Chokekijcahi, S.; Staudt, L.; Mitsuya, H.; Simpson, I.A.; Zeichner, S.L. Human immunodeficiency virus type 1 infection of H9 cells induces increased glucose transporter expression. J. Virol. 1996, 70, 7275–7279. [Google Scholar] [PubMed]
- Crompton, M. Mitochondrial intermembrane junctional complexes and their role in cell death. J. Physiol. 2000, 529, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef] [PubMed]
- Ringrose, J.H.; Jeeninga, R.E.; Berkhout, B.; Speijer, D. Proteomic studies reveal coordinated changes in T-cell expression patterns upon infection with human immunodeficiency virus type 1. J. Virol. 2008, 82, 4320–4330. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kaminiski, R.; Deshmane, S.; Langford, D.; Khalili, K.; Amini, S.; Datta, P.K. Role of hexokinase-1 in the survival of HIV-1-infected macrophages. Cell Cycle 2015, 14, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 2011, 85, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Hu, Z.Y.; Dong, X.; Tan, Z.; Li, W.; Tang, M.; Chen, L.; Yang, L.; Tao, Y.; Jiang, Y.; et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014, 33, 4568–4578. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Bonomelli, C.; Doores, K.J.; Dunlop, D.C.; Thaney, V.; Dwek, R.A.; Burton, D.R.; Crispin, M.; Scanlan, C.N. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS ONE 2011, 6, e23521. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.K.; Harvey, D.J.; Bonomelli, C.; Crispin, M.; Doores, K.J. Cell- and protein-directed glycosylation of native cleaved HIV-1 envelope. J. Virol. 2015, 89, 8932–8944. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Mulder, L.C.; Harari, A.; Simon, V. Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 5501–5506. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Booth, N.J.; Neil, S.J. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/bst-2-mediated restriction in T cells. J. Virol. 2010, 84, 12185–12199. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavanagh Williamson, M.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Taams, L.S.; Dias Zeidler, J.; Da Poian, A.T.; et al. Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1. Viruses 2018, 10, 114. https://doi.org/10.3390/v10030114
Kavanagh Williamson M, Coombes N, Juszczak F, Athanasopoulos M, Khan MB, Eykyn TR, Srenathan U, Taams LS, Dias Zeidler J, Da Poian AT, et al. Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1. Viruses. 2018; 10(3):114. https://doi.org/10.3390/v10030114
Chicago/Turabian StyleKavanagh Williamson, Maia, Naomi Coombes, Florian Juszczak, Marios Athanasopoulos, Mariam B. Khan, Thomas R. Eykyn, Ushani Srenathan, Leonie S. Taams, Julianna Dias Zeidler, Andrea T. Da Poian, and et al. 2018. "Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1" Viruses 10, no. 3: 114. https://doi.org/10.3390/v10030114
APA StyleKavanagh Williamson, M., Coombes, N., Juszczak, F., Athanasopoulos, M., Khan, M. B., Eykyn, T. R., Srenathan, U., Taams, L. S., Dias Zeidler, J., Da Poian, A. T., & Huthoff, H. (2018). Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1. Viruses, 10(3), 114. https://doi.org/10.3390/v10030114