Beet Necrotic Yellow Vein Virus Noncoding RNA Production Depends on a 5′→3′ Xrn Exoribonuclease Activity


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Yeast Transformation
In Vitro Transcription, Plant Infection, Protein, and RNA Extractions
2.3. Virus-Induced Gene Silencing in Nicotiana benthamiana or Gene Silencing Experiments in Nicotiana benthamiana
2.4. Reverse Transcription qPCR
2.5. In Vitro Xrn1 Assays
2.6. Cell-Free Xrn1 RNA Decay Assays
3. Results
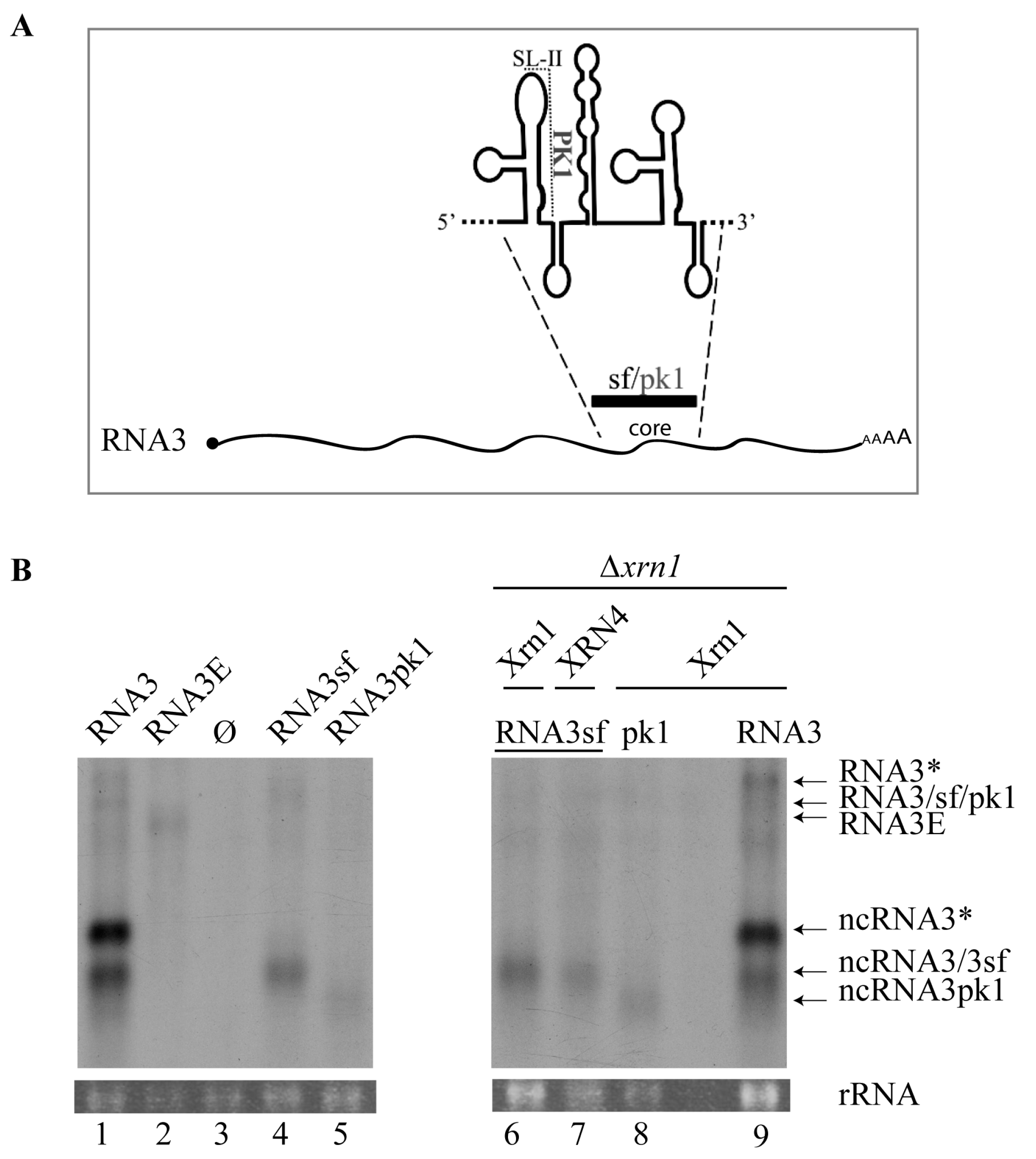
3.1. Expression of BNYVV-RNA3 in Yeast Leads to ncRNA3 Accumulation
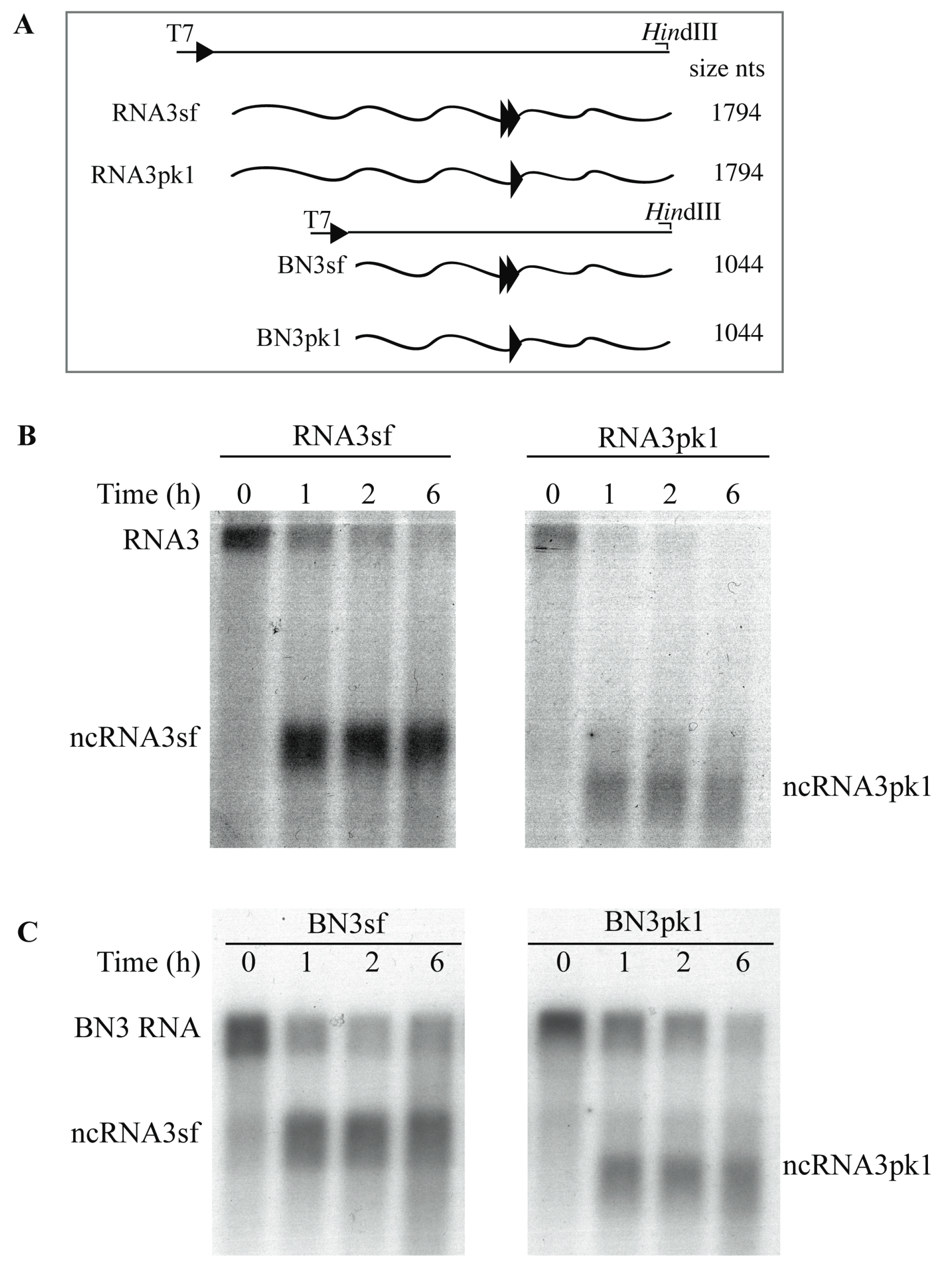
3.2. 5′→3′ Exoribonuclease Activity is Responsible for ncRNA3 Accumulation in Yeast
3.3. Substitution of the BNYVV-RNA3 Core Sequence by the Xrn1-Resistant West Nile Virus Core Sequence Produces a New ncRNA3 in S. cerevisiae Strains Expressing Xrn1 or Arabidopsis thaliana XRN4
3.4. Xrn1 Produces ncRNA3 In Vitro
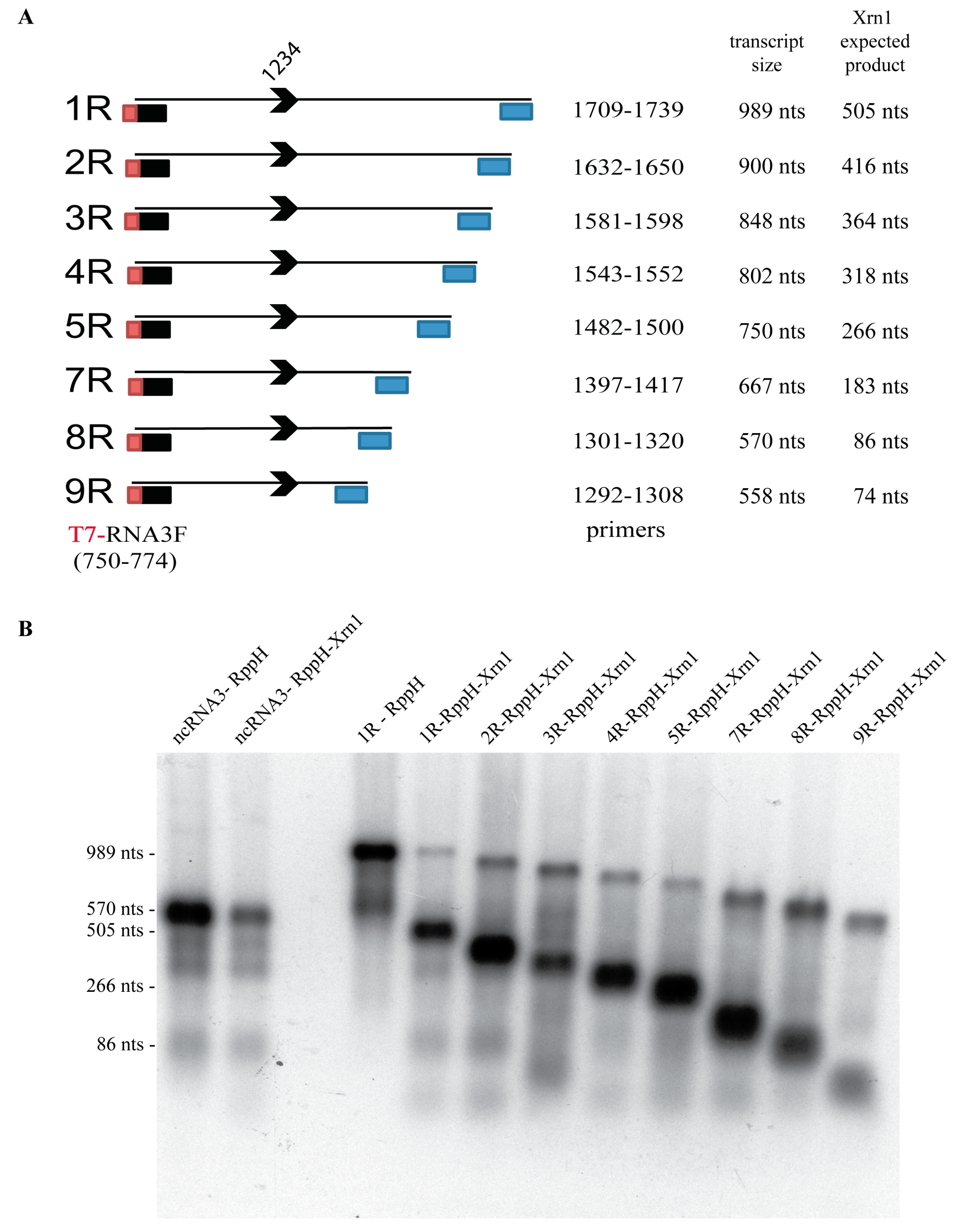
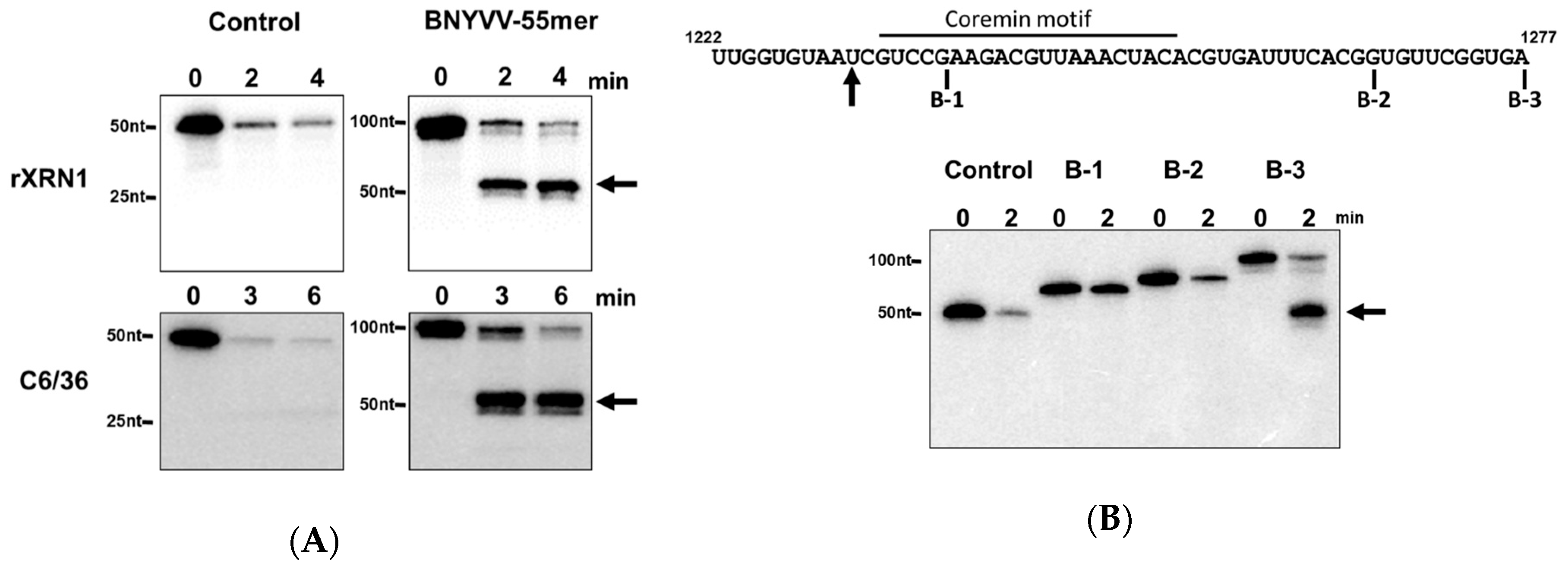
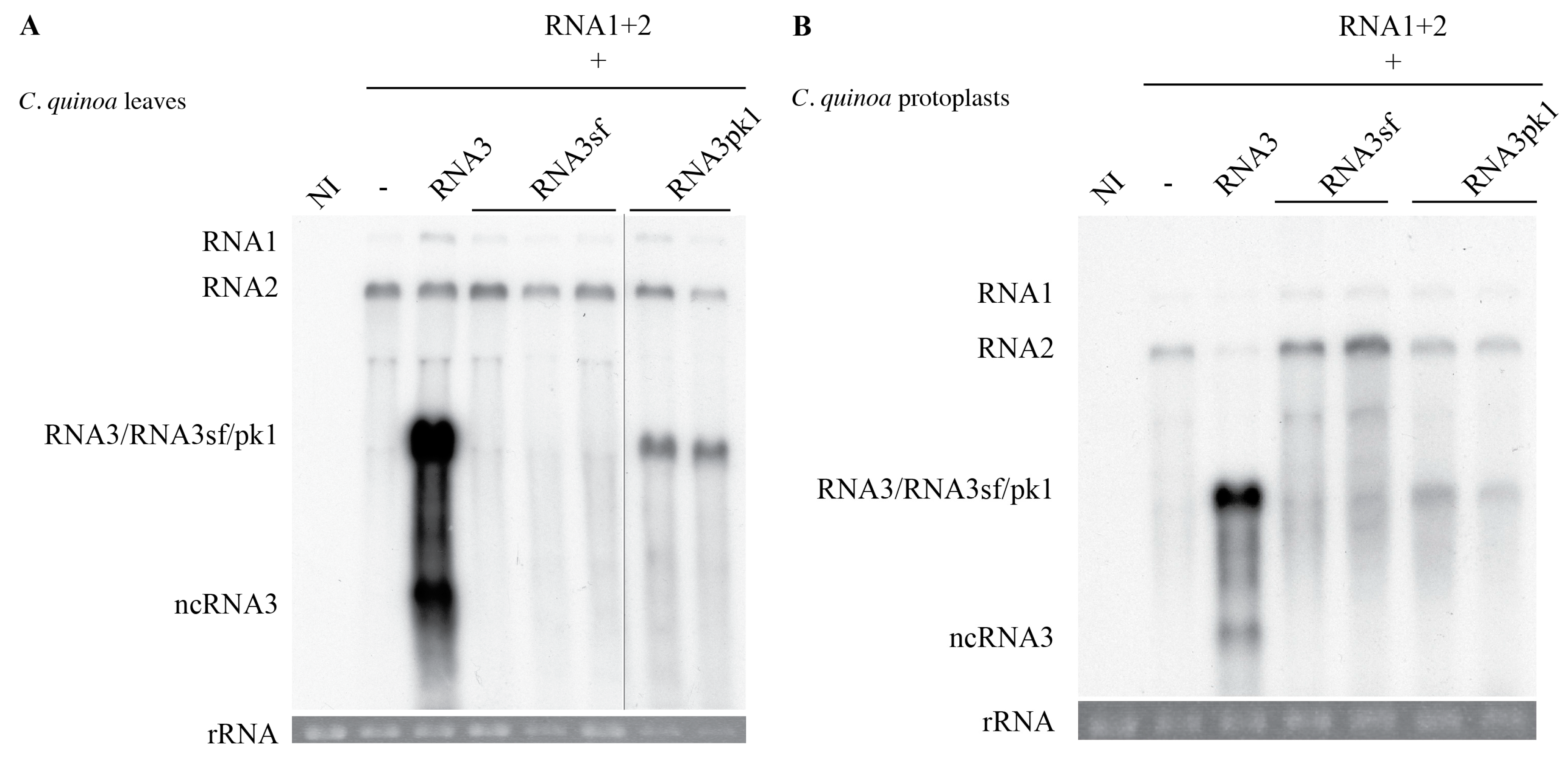
3.5. A Minimal Sequence of 43 nt within the ncRNA Sequence is Sufficient for Xrn1 Stalling
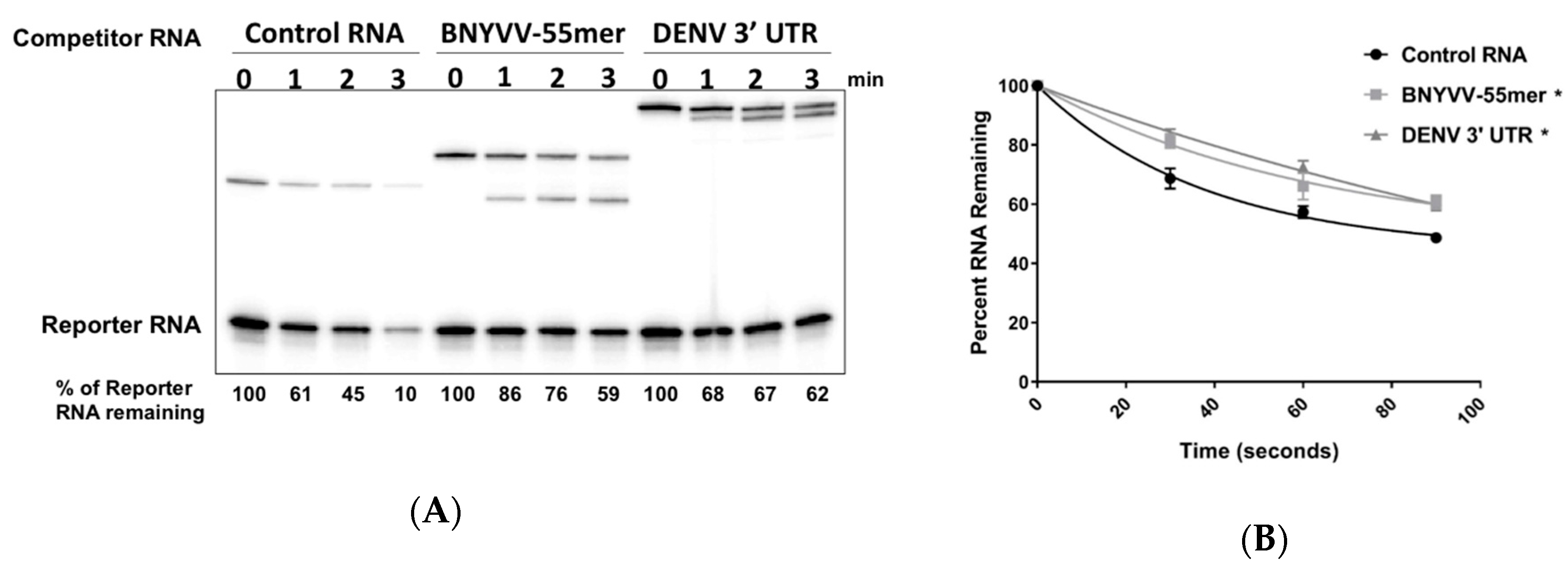
3.6. Accumulation of BNYVV ncRNA Represses Xrn Activity In Vitro
3.7. Exoribonuclease Stalling is Not Sufficient to Induce the Systemic Spread of the BNYVV in Beta macrocarpa
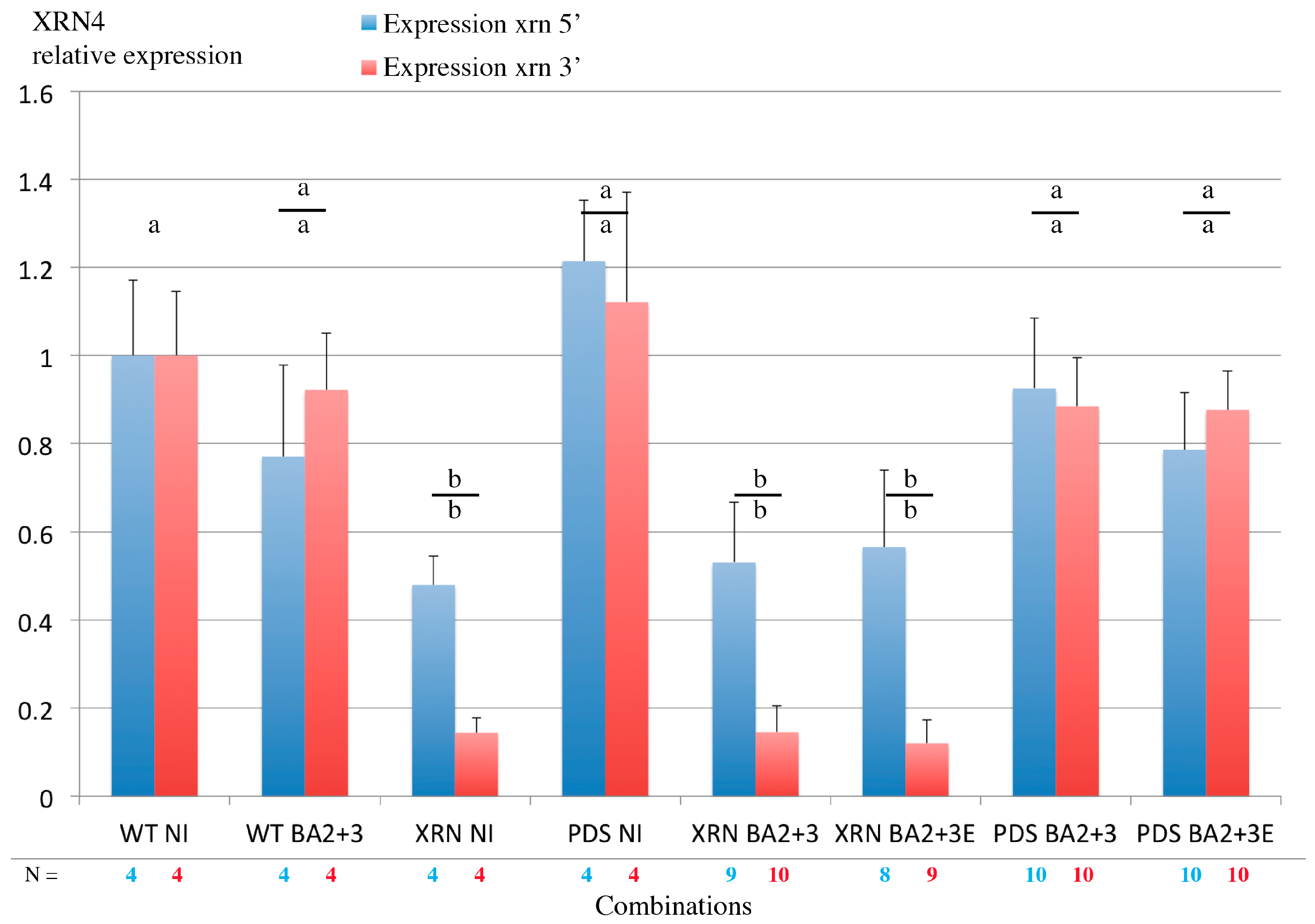
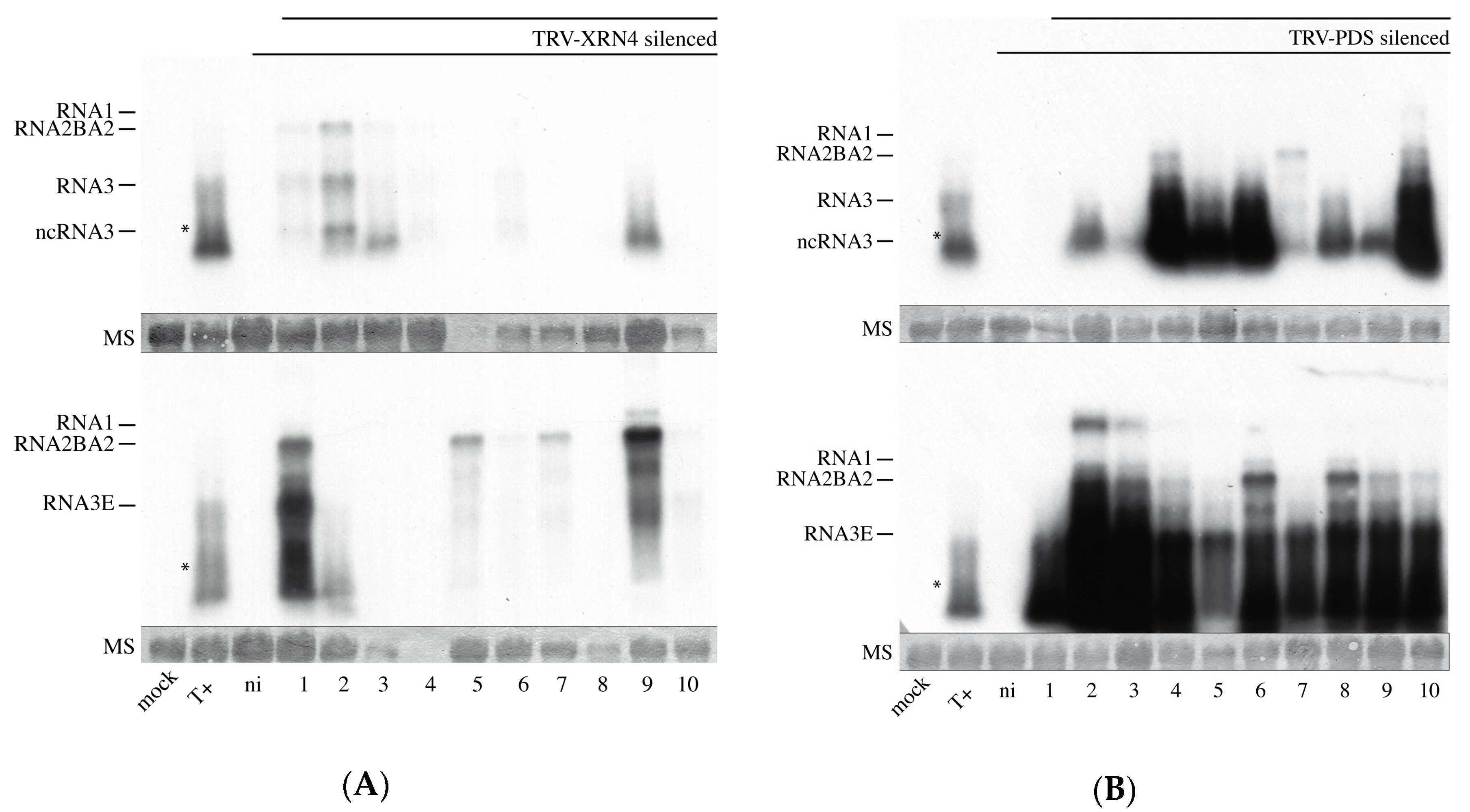
3.8. Virus-Induced Gene Silencing of XRN4 Exoribonuclease Affects Viral Accumulation and Does Not Allow Systemic Spread in N. benthamiana
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Łabno, A.; Tomecki, R.; Dziembowski, A. Cytoplasmic RNA decay pathways—Enzymes and mechanisms. Biochim. Biophys. Acta 2016, 1863, 3125–3147. [Google Scholar] [CrossRef] [PubMed]
- Rozovics, J.M.; Chase, A.J.; Cathcart, A.L.; Chou, W.; Gershon, P.D.; Palusa, S.; Wilusz, J.; Semler, B.L. Picornavirus modification of a host mRNA decay protein. mBio 2012, 3, e00431-12. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, K.J.; Dickson, A.M.; Chaskey, E.L.; Garneau, N.L.; Wilusz, C.J.; Wilusz, J. Sindbis virus usurps the cellular hur protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 2010, 8, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Csorba, T.; Kontra, L.; Burgyán, J. Viral silencing suppressors: Tools forged to fine-tune host-pathogen coexistence. Virology 2015, 479–480, 85–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.L.; Wilusz, J. Cytoplasmic viruses: Rage against the (cellular RNA decay) machine. PLoS Pathog. 2013, 9, e1003762. [Google Scholar] [CrossRef] [PubMed]
- Molleston, J.M.; Cherry, S. Attacked from all sides: RNA decay in antiviral defense. Viruses 2017, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Poole, T.L.; Stevens, A. Structural modifications of RNA influence the 5′ exoribonucleolytic hydrolysis by Xrn1 and Hke1 of saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1997, 235, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.A.G.C.; Pereira, C.F.; Dalebout, T.J.; Spaan, W.J.M.; Bredenbeek, P.J. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease Xrn1. J. Virol. 2010, 84, 11395–11406. [Google Scholar] [CrossRef] [PubMed]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Balmori Melian, E.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.L.; Anderson, J.R.; Kumagai, Y.; Wilusz, C.J.; Akira, S.; Khromykh, A.A.; Wilusz, J. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease Xrn1 and alters host mRNA stability. RNA 2012, 18, 2029–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, E.G.; Costantino, D.A.; Rabe, J.L.; Moon, S.L.; Wilusz, J.; Nix, J.C.; Kieft, J.S. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science 2014, 344, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds trim25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Göertz, G.P.; Fros, J.J.; Miesen, P.; Vogels, C.B.F.; van der Bent, M.L.; Geertsema, C.; Koenraadt, C.J.M.; van Rij, R.P.; van Oers, M.M.; Pijlman, G.P. Noncoding subgenomic flavivirus RNA is processed by the mosquito RNA interference machinery and determines west nile virus transmission by culex pipiens mosquitoes. J. Virol. 2016, 90, 10145–10159. [Google Scholar] [CrossRef] [PubMed]
- Schnettler, E.; Sterken, M.G.; Leung, J.Y.; Metz, S.W.; Geertsema, C.; Goldbach, R.W.; Vlak, J.M.; Kohl, A.; Khromykh, A.A.; Pijlman, G.P. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and mammalian cells. J. Virol. 2012, 86, 13486–13500. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.L.; Dodd, B.J.T.; Brackney, D.E.; Wilusz, C.J.; Ebel, G.D.; Wilusz, J. Flavivirus sfRNA suppresses antiviral RNA interference in cultured cells and mosquitoes and directly interacts with the RNAi machinery. Virology 2015, 485, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.G.; Moon, S.L.; Wilusz, J.; Kieft, J.S. RNA structures that resist degradation by Xrn1 produce a pathogenic dengue virus RNA. eLife 2014, 3, e01892. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.O.; Mizumoto, H.; Nagano, H.; Imoto, Y.; Takigawa, K.; Sarawaneeyaruk, S.; Kaido, M.; Mise, K.; Okuno, T. A viral noncoding RNA generated by cis-element-mediated protection against 5′→3′ RNA decay represses both cap-independent and cap-dependent translation. J. Virol. 2008, 82, 10162–10174. [Google Scholar] [CrossRef] [PubMed]
- Pashler, A.L.; Towler, B.P.; Jones, C.I.; Newbury, S.F. The roles of the exoribonucleases Dis3l2 and Xrn1 in human disease. Biochem. Soc. Trans. 2016, 44, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Adjibade, P.; Mazroui, R. Control of mRNA turnover: Implication of cytoplasmic RNA granules. Semin. Cell Dev. Biol. 2014, 34, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Green, P.J. mRNA degradation machinery in plants. J. Plant Biol. 2009, 52, 114–124. [Google Scholar] [CrossRef]
- Cheng, C.P.; Jaag, H.M.; Jonczyk, M.; Serviene, E.; Nagy, P.D. Expression of the arabidopsis Xrn4p 5′-3′ exoribonuclease facilitates degradation of tombusvirus RNA and promotes rapid emergence of viral variants in plants. Virology 2007, 368, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yang, J.; Yan, F.; Lu, Y.; Jiang, S.; Lin, L.; Zheng, H.; Chen, H.; Chen, J. Silencing of nbxrn4 facilitates the systemic infection of tobacco mosaic virus in nicotiana benthamiana. Virus Res. 2011, 158, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Lin, T.L.; Lin, J.W.; Han, Y.T.; Huang, Y.T.; Hsu, Y.H.; Meng, M. Promotion of bamboo mosaic virus accumulation in nicotiana benthamiana by 5′→3′ exonuclease nbxrn4. Front. Microbiol. 2015, 6, 1508. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, D.; Ratti, C. Benyvirus. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 1133–1138. [Google Scholar]
- Gilmer, D.; Ratti, C.; Consortium, I.R. Ictv virus taxonomy profile: Benyviridae. J. Gen. Virol. 2017, 98, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Quillet, L.; Guilley, H.; Jonard, G.; Richards, K. In vitro synthesis of biologically active beet necrotic yellow vein virus RNA. Virology 1989, 172, 293–301. [Google Scholar] [PubMed]
- Chiba, S.; Hleibieh, K.; Delbianco, A.; Klein, E.; Ratti, C.; Ziegler-Graff, V.; Bouzoubaa, S.; Gilmer, D. The benyvirus RNA silencing suppressor is essential for long-distance movement, requires both zinc-finger and nols basic residues but not a nucleolar localization for its silencing-suppression activity. Mol. Plant-Microbe Interact. 2013, 26, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Flobinus, A.; Hleibieh, K.; Klein, E.; Ratti, C.; Bouzoubaa, S.; Gilmer, D. A viral noncoding RNA complements a weakened viral RNA silencing suppressor and promotes efficient systemic host infection. Viruses 2016, 8, 272. [Google Scholar] [CrossRef] [PubMed]
- Balmori, E.; Gilmer, D.; Richards, K.; Guilley, H.; Jonard, G. Mapping the promoter for subgenomic RNA synthesis on beet necrotic yellow vein virus RNA 3. Biochimie 1993, 75, 517–521. [Google Scholar] [CrossRef]
- Peltier, C.; Klein, E.; Hleibieh, K.; D’Alonzo, M.; Hammann, P.; Bouzoubaa, S.; Ratti, C.; Gilmer, D. Beet necrotic yellow vein virus subgenomic RNA3 is a cleavage product leading to stable non-coding RNA required for long-distance movement. J. Gen. Virol. 2012, 93, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Lauber, E.; Guilley, H.; Tamada, T.; Richards, K.E.; Jonard, G. Vascular movement of beet necrotic yellow vein virus in beta macrocarpa is probably dependent on an RNA 3 sequence domain rather than a gene product. J. Gen. Virol. 1998, 79, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Ratti, C.; Hleibieh, K.; Bianchi, L.; Schirmer, A.; Autonell, C.R.; Gilmer, D. Beet soil-borne mosaic virus RNA-3 is replicated and encapsidated in the presence of bnyvv RNA-1 and -2 and allows long distance movement in beta macrocarpa. Virology 2009, 385, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, D.; Richards, K.; Jonard, G.; Guilley, H. Cis-active sequences near the 5′-termini of beet necrotic yellow vein virus RNAs 3 and 4. Virology 1992, 190, 55–67. [Google Scholar] [CrossRef]
- Moon, S.L.; Blackinton, J.G.; Anderson, J.R.; Dozier, M.K.; Dodd, B.J.T.; Keene, J.D.; Wilusz, C.J.; Bradrick, S.S.; Wilusz, J. Xrn1 stalling in the 5′ utr of hepatitis c virus and bovine viral diarrhea virus is associated with dysregulated host mRNA stability. PLoS Pathog. 2015, 11, e1004708. [Google Scholar] [CrossRef] [PubMed]
- Sinturel, F.; Brechemier-Baey, D.; Kiledjian, M.; Condon, C.; Benard, L. Activation of 5′-3′ exoribonuclease Xrn1 by cofactor Dcs1 is essential for mitochondrial function in yeast. Proc. Natl. Acad. Sci. USA 2012, 109, 8264–8269. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/peg method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Guilley, H.; Bortolamiol, D.; Jonard, G.; Bouzoubaa, S.; Ziegler-Graff, V. Rapid screening of RNA silencing suppressors by using a recombinant virus derived from beet necrotic yellow vein virus. J. Gen. Virol. 2009, 90, 2536–2541. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Link, D.; Schirmer, A.; Erhardt, M.; Gilmer, D. Sequence variation within beet necrotic yellow vein virus p25 protein influences its oligomerization and isolate pathogenicity on tetragonia expansa. Virus Res. 2007, 126, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Veidt, I.; Bouzoubaa, S.E.; Leiser, R.M.; Ziegler-Graff, V.; Guilley, H.; Richards, K.; Jonard, G. Synthesis of full-length transcripts of beet western yellows virus RNA: Messenger properties and biological activity in protoplasts. Virology 1992, 186, 192–200. [Google Scholar] [CrossRef]
- Jupin, I.; Richards, K.; Jonard, G.; Guilley, H.; Pleij, C.W. Mapping sequences required for productive replication of beet necrotic yellow vein virus RNA 3. Virology 1990, 178, 273–280. [Google Scholar] [CrossRef]
- Delbianco, A.; Lanzoni, C.; Klein, E.; Rubies Autonell, C.; Gilmer, D.; Ratti, C. Agroinoculation of Beet necrotic yellow vein virus cDNA clones results in plant systemic infection and efficient Polymyxa betae transmission. Mol. Plant Pathol. 2013, 14, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, K.; Anderson, J.R.; Wilusz, J. Development of an in vitro mRNA decay system in insect cells. In Post-Transcriptional Gene Regulation; Wilusz, J., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 277–288. [Google Scholar]
- Mukherjee, D.; Gao, M.; O’Connor, J.P.; Raijmakers, R.; Pruijn, G.; Lutz, C.S.; Wilusz, J. The mammalian exosome mediates the efficient degradation of mRNAs that contain au-rich elements. EMBO J. 2002, 21, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Xiang, S.; Xiang, K.; Manley, J.L.; Tong, L. Structural and biochemical studies of the 5′→3′ exoribonuclease Xrn1. Nat. Struct. Mol. Biol. 2011, 18, 270. [Google Scholar] [CrossRef] [PubMed]
- Balk, B.; Maicher, A.; Dees, M.; Klermund, J.; Luke-Glaser, S.; Bender, K.; Luke, B. Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat. Struct. Mol. Biol. 2013, 20, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Van Hoof, A.; Lennertz, P.; Parker, R. Three conserved members of the RNase d family have unique and overlapping functions in the processing of 5S, 5.8S, U4, U5, RNase MRP and RNase P RNAs in yeast. EMBO J. 2000, 19, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Feigenbutz, M.; Garland, W.; Turner, M.; Mitchell, P. The exosome cofactor Rrp47 is critical for the stability and normal expression of its associated exoribonuclease Rrp6 in saccharomyces cerevisiae. PLoS ONE 2013, 8, e80752. [Google Scholar] [CrossRef] [PubMed]
- Kenna, M.; Stevens, A.; McCammon, M.; Douglas, M.G. An essential yeast gene with homology to the exonuclease-encoding Xrn1/Kem1 gene also encodes a protein with exoribonuclease activity. Mol. Cell Biol. 1993, 13, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Harigaya, Y.; Parker, R. Global analysis of mRNA decay intermediates in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2012, 109, 11764–11769. [Google Scholar] [CrossRef] [PubMed]
- Charley, P.A.; Wilusz, C.J.; Wilusz, J. Identification of phlebovirus and arenavirus RNA sequences that stall and repress the exoribonuclease Xrn1. J. Biol. Chem. 2018, 293, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Ratcliff, F.; Martin-Hernandez, A.M.; Baulcombe, D.C. Technical advance. Tobacco rattle virus as a vector for analysis of gene function by silencing. Plant J. 2001, 25, 237–245. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combinations | 1st Experiment | 2nd Experiment |
|---|---|---|
| RNA1+2 | 0% (5) | 0% (5) |
| RNA1+2+3 | 70% (10) | 90% (10) |
| RNA1+2+3E | 0% (10) | NT |
| RNA1+2+3sf † | 0% (10) | 0% (10) |
| RNA1+2+3pk1 | 0% (10) | 0% (10) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flobinus, A.; Chevigny, N.; Charley, P.A.; Seissler, T.; Klein, E.; Bleykasten-Grosshans, C.; Ratti, C.; Bouzoubaa, S.; Wilusz, J.; Gilmer, D. Beet Necrotic Yellow Vein Virus Noncoding RNA Production Depends on a 5′→3′ Xrn Exoribonuclease Activity. Viruses 2018, 10, 137. https://doi.org/10.3390/v10030137
Flobinus A, Chevigny N, Charley PA, Seissler T, Klein E, Bleykasten-Grosshans C, Ratti C, Bouzoubaa S, Wilusz J, Gilmer D. Beet Necrotic Yellow Vein Virus Noncoding RNA Production Depends on a 5′→3′ Xrn Exoribonuclease Activity. Viruses. 2018; 10(3):137. https://doi.org/10.3390/v10030137
Chicago/Turabian StyleFlobinus, Alyssa, Nicolas Chevigny, Phillida A. Charley, Tanja Seissler, Elodie Klein, Claudine Bleykasten-Grosshans, Claudio Ratti, Salah Bouzoubaa, Jeffrey Wilusz, and David Gilmer. 2018. "Beet Necrotic Yellow Vein Virus Noncoding RNA Production Depends on a 5′→3′ Xrn Exoribonuclease Activity" Viruses 10, no. 3: 137. https://doi.org/10.3390/v10030137
APA StyleFlobinus, A., Chevigny, N., Charley, P. A., Seissler, T., Klein, E., Bleykasten-Grosshans, C., Ratti, C., Bouzoubaa, S., Wilusz, J., & Gilmer, D. (2018). Beet Necrotic Yellow Vein Virus Noncoding RNA Production Depends on a 5′→3′ Xrn Exoribonuclease Activity. Viruses, 10(3), 137. https://doi.org/10.3390/v10030137