Molecular Characterization of a Novel Species of Capillovirus from Japanese Apricot (Prunus mume)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Viral Source, and Maintenance
2.2. Determination of the Herbaceous Host Range of PM14 Isolate
2.3. Analysis of Double-Stranded RNAs by High-Throughput Sequencing and Completion of the PM14 Isolate Genome Sequence
2.4. Total Nucleic Acid Extraction and RT-PCR Detection of Mume Virus A
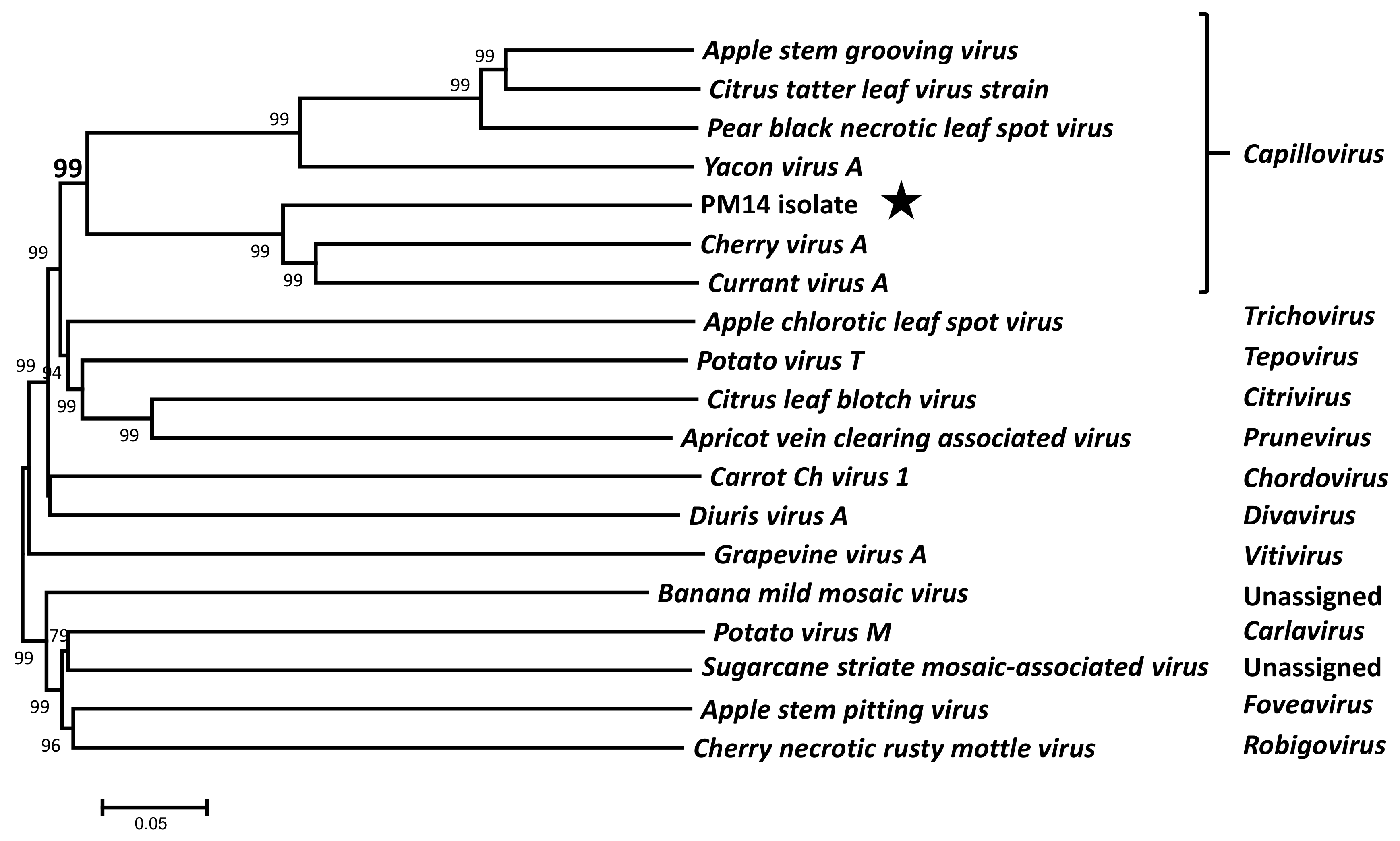
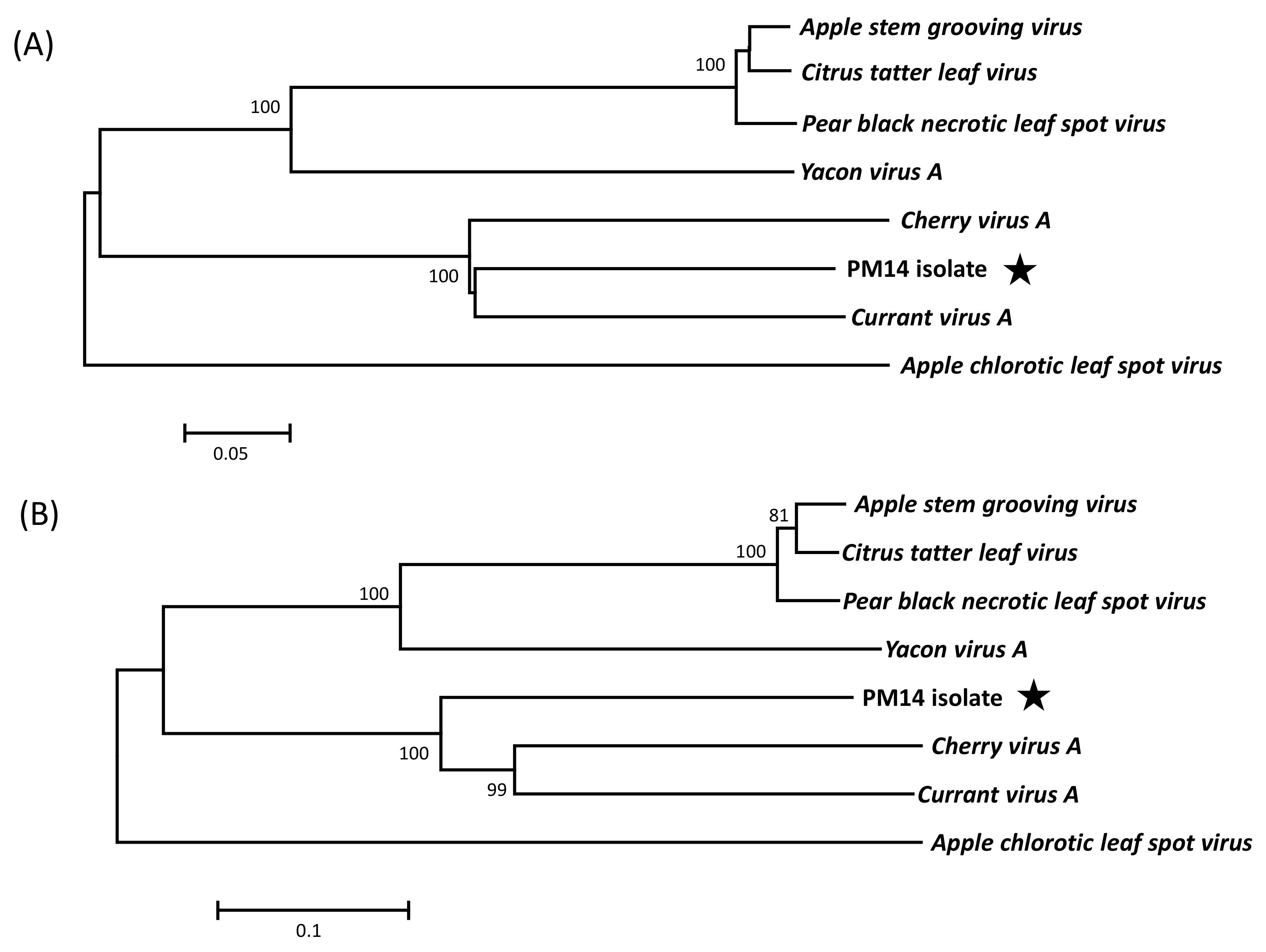
2.5. Phylogenetic Analyses
3. Results and Discussion
3.1. Illumina Sequencing of Double-Stranded RNAs Purified from the PM14 Japanese Apricot Source
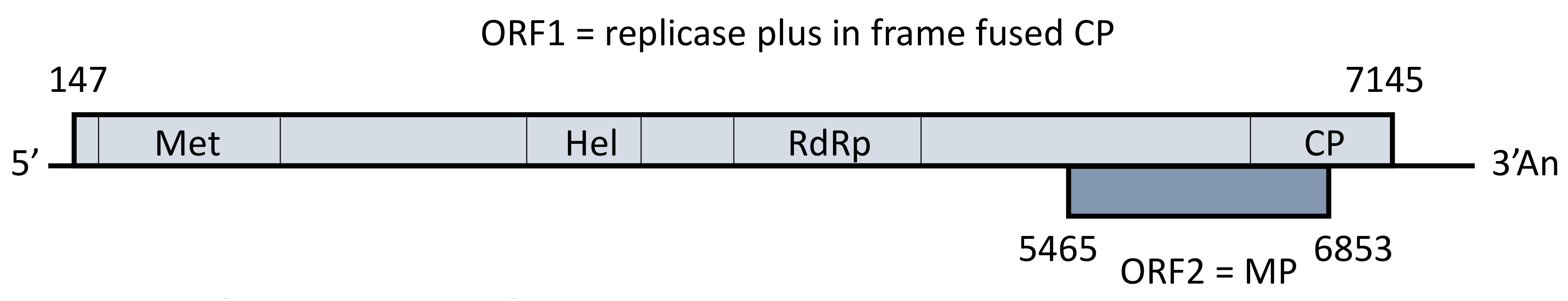
3.2. Genome Organization and Phylogenetic Relationships of PM14 Isolate
3.3. Biological Characterization and Incidence of Mume virus A
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.; Martínez-Gómez, P.; Marais, A.; Sánchez-Navarro, J.A.; Pallás, V.; Candresse, T. Recent advances and prospects in Prunus virology. Ann. Appl. Biol. 2017, 171, 125–138. [Google Scholar] [CrossRef]
- Marais, A.; Faure, C.; Mustafayev, E.; Candresse, T. Characterization of new isolates of Apricot vein clearing-associated virus and of a new Prunus-infecting virus: Evidence for recombination as a driving force in Betaflexiviridae evolution. PLoS ONE 2015, 10, e0129469. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.; Susaimuthu, J.; Eastwell, K.C. Genomic analyses of Cherry rusty mottle group and Cherry twisted leaf-associated viruses reveal a possible new genus within the family Betaflexiviridae. Phytopathology 2015, 105, 399–408. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cai, L.; Zhou, L.; Yang, Z.; Hong, N.; Wang, G.; Li, S.; Xu, W. Deep sequencing reveals the first fabavirus infecting peach. Sci. Rep. 2017, 7, 11329. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.; Mekuria, T.A.; Pillai, S.S.; Eastwell, K.C. High-throughput sequencing identifies novel viruses in nectarine: Insights to the etiology of stem-pitting disease. Phytopathology 2016, 106, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Igori, D.; Lim, S.; Baek, D.; Kim, S.Y.; Seo, E.; Cho, I.-S.; Choi, G.-S.; Lim, H.-S.; Moon, J.S. Complete nucleotide sequence and genome organization of peach virus D, a putative new member of the genus Marafivirus. Arch. Virol. 2017, 162, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Faure, C.; Couture, C.; Bergey, B.; Gentit, P.; Candresse, T. Characterization by deep sequencing of divergent Plum bark necrosis stem pitting-associated virus isolates and development of a broad-spectrum PBNSPaV detection assay. Phytopathology 2014, 104, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Candresse, T.; Faure, C.; Theil, S.; Marais, A. First report of Nectarine stem pitting-associated virus infecting Prunus mume in Japan. Plant Dis. 2017, 101, 393. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Alabi, O.J.; Westrick, N.M.; Golin, D. Prunus geminivirus A: A novel Grablovirus infecting Prunus spp. Plant Dis. 2017. [Google Scholar] [CrossRef]
- Jo, Y.; Lian, S.; Cho, J.K.; Chu, H.; Cho, W.K. First Report of Hop stunt viroid in Japanese Apricot (Prunus mume) in Korea. Plant Dis. 2017, 101, 1332. [Google Scholar] [CrossRef]
- Massart, S.; Jijakli, M.M.; Kummert, J. Apple stem grooving virus. In Virus and Virus-Like Diseases of Pome and Stone Fruits; Hadidi, A., Barba, M., Candresse, T., Jelkmann, W., Eds.; American Phytopathological Society Press: St. Paul, MN, USA, 2011; pp. 29–33. [Google Scholar]
- Nemchinov, L.G.; Gentit, G.; Zemcic, E.; Candresse, T.; Hadidi, A. Apricot latent virus. In Virus and Virus-Like Diseases of Pome and Stone Fruits; Hadidi, A., Barba, M., Candresse, T., Jelkmann, W., Eds.; American Phytopathological Society Press: St. Paul, MN, USA, 2011; pp. 97–101. [Google Scholar]
- Gentit, P.; Foissac, X.; Svanella-Dumas, L.; Peypelut, M.; Candresse, T. Characterization of two different Apricot latent virus variants associated with peach asteroid spot and peach sooty ringspot diseases. Arch. Virol. 2001, 146, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Candresse, T.; Marais, A.; Faure, C.; Gentit, P. Association of Little cherry virus 1 with the Shirofugen stunt disease and characterization of the genome of a divergent LChV1 isolate. Phytopathology 2013, 103, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Youssef, F.; Marais, A.; Faure, C.; Barone, M.; Gentit, P.; Candresse, T. Characterization of Prunus-infecting Apricot latent virus-like Foveaviruses: Evolutionary and taxonomic implications. Virus Res. 2011, 155, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Foissac, X.; Svanella-Dumas, L.; Gentit, P.; Dulucq, M.J.; Marais, A.; Candresse, T. Polyvalent degenerate oligonucleotides reverse transcription-polymerase chain reaction: A polyvalent detection and characterization tool for Trichoviruses, Capilloviruses, and Foveaviruses. Phytopathology 2005, 95, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTALW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap, penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 3, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Petrzik, K.; Pribylova, J.; Koloniuk, I.; Spak, J. Molecular characterization of a novel capillovirus from red currant. Arch. Virol. 2016, 161, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Candresse, T.; Hammond, J.; Kreuze, J.F.; Martelli, G.P.; Namba, S.; Pearson, M.N.; Ryu, K.H.; Saldarelli, P.; Yoshikawa, N. Family Betaflexiviridae. In Virus Taxonomy-Ninth Report on the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: Cambridge, MA, USA, 2012; pp. 920–941. [Google Scholar]
- Yoshikawa, N.; Imaizumi, M.; Takahashi, T.; Inouye, N. Striking similarities between the nucleotide sequence and genome organization of citrus tatter leaf and apple stem grooving capilloviruses. J. Gen. Virol. 1993, 74, 2743–2747. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Min, Y.; Hong, S.; Kwon, M.; Kim, D.; Kim, H.; Choi, Y.; Lee, S.; Yang, J. Nucleotide sequences of a Korean isolate of Apple stem grooving virus associated with black necrotic leaf spot disease on pear (Pyrus pyrifolia). Mol. Cells 2004, 18, 192–199. [Google Scholar] [PubMed]
- Marais, A.; Candresse, T.; Svanella-Dumas, L.; Jelkmann, W. Cherry virus A. In Virus and Virus-Like Diseases of Pome and Stone Fruits; Hadidi, A., Barba, M., Candresse, T., Jelkmann, W., Eds.; APS Press: St. Paul, MN, USA, 2011; pp. 147–150. [Google Scholar]
- Desvignes, J.C.; Boye, R.; Cornaggia, D.; Grasseau, N. Apple stem grooving virus. In Virus Diseases of Fruit Trees; Hassan, M., Myrta, A., Polák, J., Eds.; Centre Technique Interprofessionnel des Fruits et Légumes (CTIFL): Paris, France, 1999; pp. 155–156. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′-3′) | Genome Coordinates | Amplicon Size (nt) |
|---|---|---|---|
| Capillo-mume-5Race2 # | CCTTGCATGGTTGTTGTTGAAGTCCTCCC | 251–223 | 252 |
| Capillo-mume-F1 | AACAACAACCATGCAAGGTTTGAG | 234–257 | 503 |
| Capillo-mume-R1 | GCTAGAACACACTTAGGCCGCAA | 736–714 | |
| Capillo-mume-F2 | GGAATGTTGATACATACAGACA | 1629–1650 | 742 |
| Capillo-mume-R2 | CGTCTGAGCCTAATCCATACAC | 2370–2349 | |
| Capillo-mume-F3 | TGGATTTATTGAACTTCTCATAC | 2831–2853 | 406 |
| Capillo-mume-R3 | CGTCACAATCACACCAAATCTG | 3236–3215 | |
| Capillo-mume-F6 | GATGTACGAGGATTCAGTGG | 4034–4053 | 451 |
| Capillo-mume-R6 | AATGAGGGAGTTAGAAACACC | 4484–4464 | |
| Capillo-mume-R7 ~ | ACCAACTGTTATGACAGATTC | 5117–5097 | 1084 |
| LD-PolyT @ | CACTGGCGGCCGCTCGAGCATGTAC(T)25NN | ||
| Capillo-mume-LD | GAGCACCATTGGAGGGTGTGT | 7462–7482 | 183 |
| LD-Prime | CACTGGCGGCCGCTCGAGCATGTAC |
| Virus @ | Percent Amino Acid Identity (Size [aa]) | ||
|---|---|---|---|
| Replicase (2105) | Coat Protein (228) | Movement Protein (463) | |
| CuVA | 53.7% (2296) | 65.4% (229) | 53.3% (462) |
| CVA | 54.4% (2112) | 62.7% (232) | 53.7% (463) |
| ASGV | 25.7% (1868) | 32.1% (238) | 29.1% (321) |
| YVA | 24.6% (1868) | 30.2% (215) | 26.2% (324) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marais, A.; Faure, C.; Theil, S.; Candresse, T. Molecular Characterization of a Novel Species of Capillovirus from Japanese Apricot (Prunus mume). Viruses 2018, 10, 144. https://doi.org/10.3390/v10040144
Marais A, Faure C, Theil S, Candresse T. Molecular Characterization of a Novel Species of Capillovirus from Japanese Apricot (Prunus mume). Viruses. 2018; 10(4):144. https://doi.org/10.3390/v10040144
Chicago/Turabian StyleMarais, Armelle, Chantal Faure, Sébastien Theil, and Thierry Candresse. 2018. "Molecular Characterization of a Novel Species of Capillovirus from Japanese Apricot (Prunus mume)" Viruses 10, no. 4: 144. https://doi.org/10.3390/v10040144
APA StyleMarais, A., Faure, C., Theil, S., & Candresse, T. (2018). Molecular Characterization of a Novel Species of Capillovirus from Japanese Apricot (Prunus mume). Viruses, 10(4), 144. https://doi.org/10.3390/v10040144