Full-Genome Characterization and Genetic Evolution of West African Isolates of Bagaza Virus

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Primers Design
2.2. Virus stock preparation and RNA extraction
2.3. RT-PCR and Sequencing
2.4. Sequence Properties Analysis
2.5. Prediction of N-Glycosylation Sites
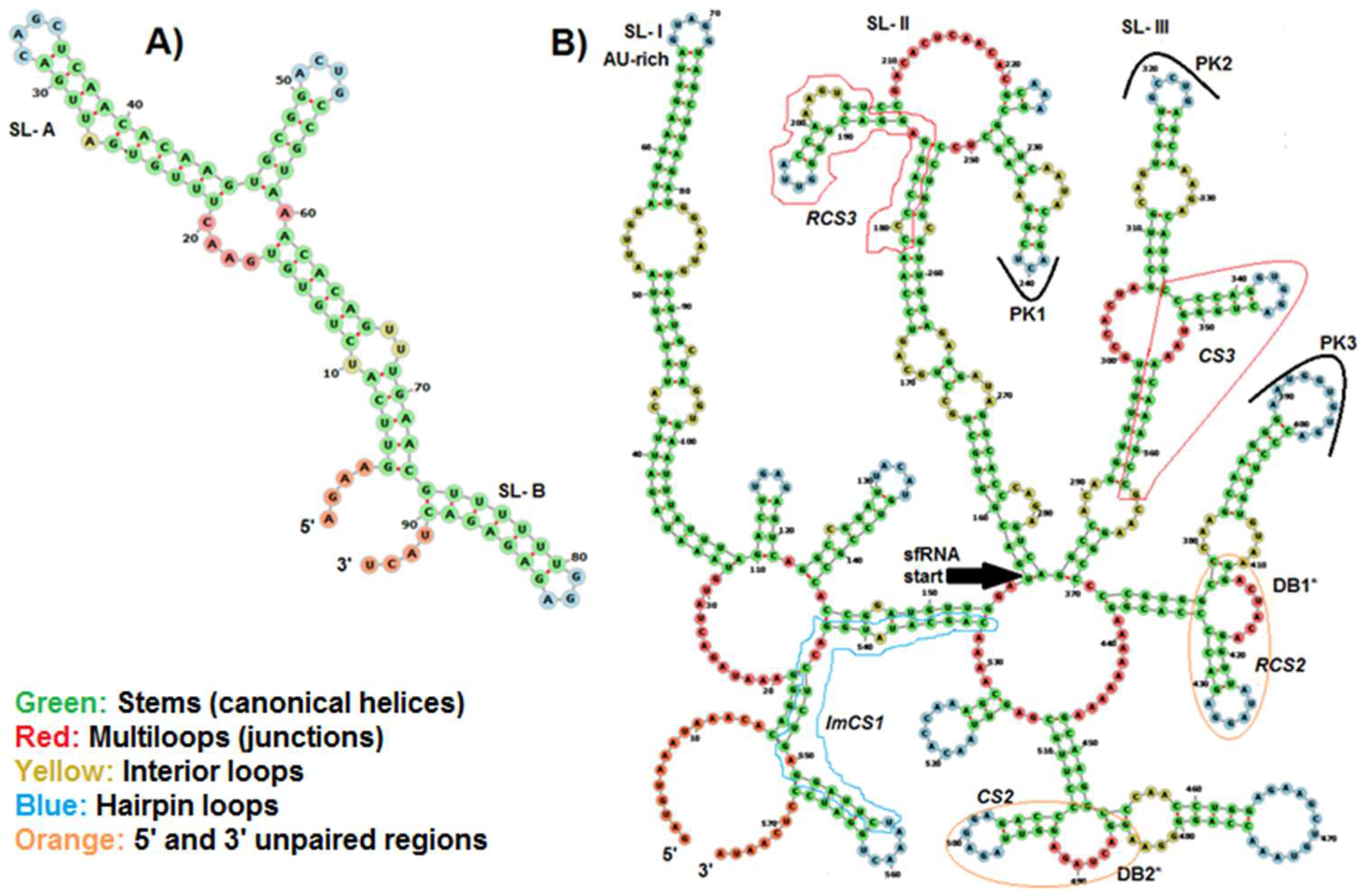
2.6. Prediction of Conserved Structural RNA Domains in 5′ and 3′ UTRs
2.7. Phylogenetic Tree Inference
2.8. Recombination Detection
2.9. Evaluation of Selection Patterns on ORFs
2.10. Bayesian Analysis
2.11. Codon Adaptation Indexes to Human House-Keeping Genes
3. Results
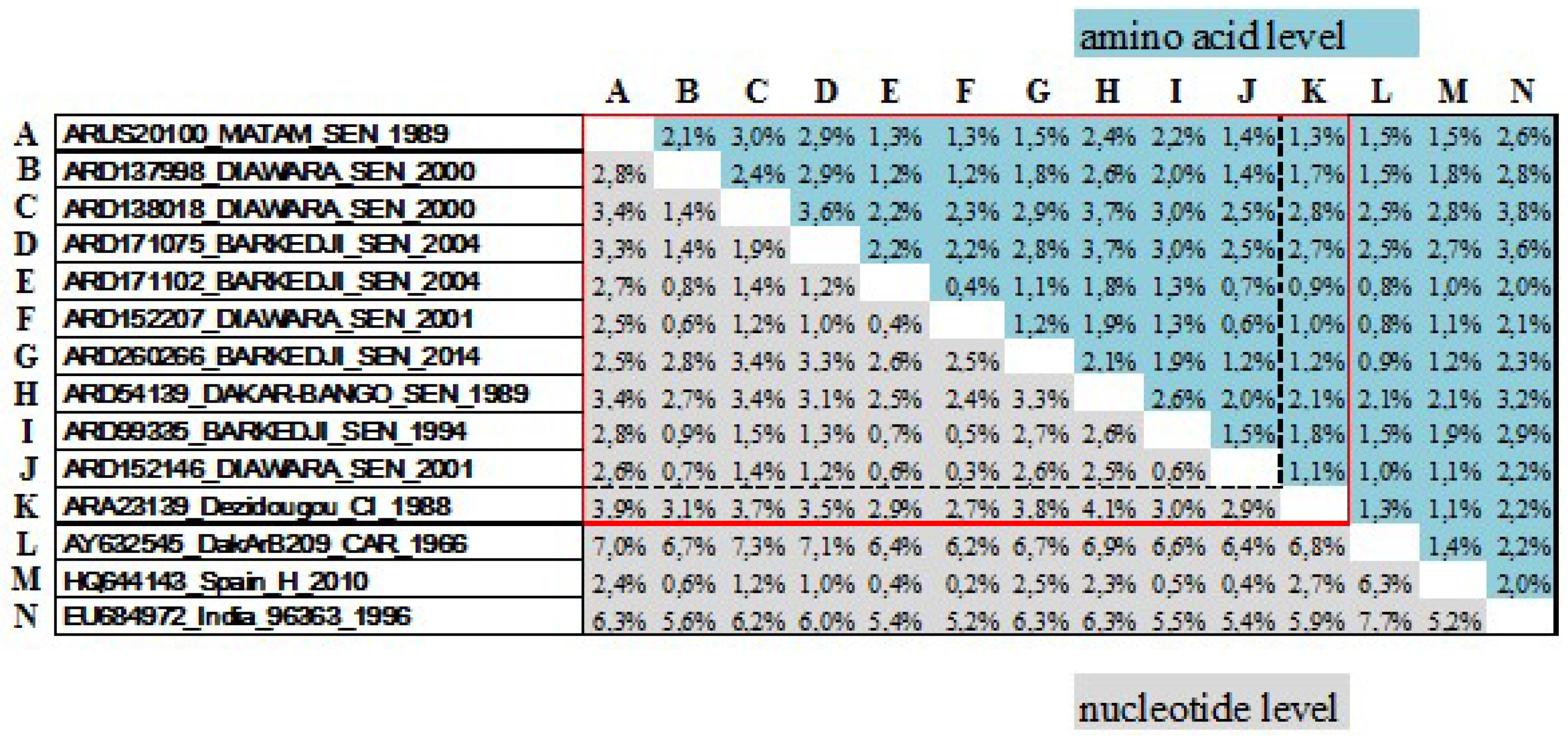
3.1. Genetic Diversity
3.2. Genetic Motifs and Informative Sites on BAGV Genome
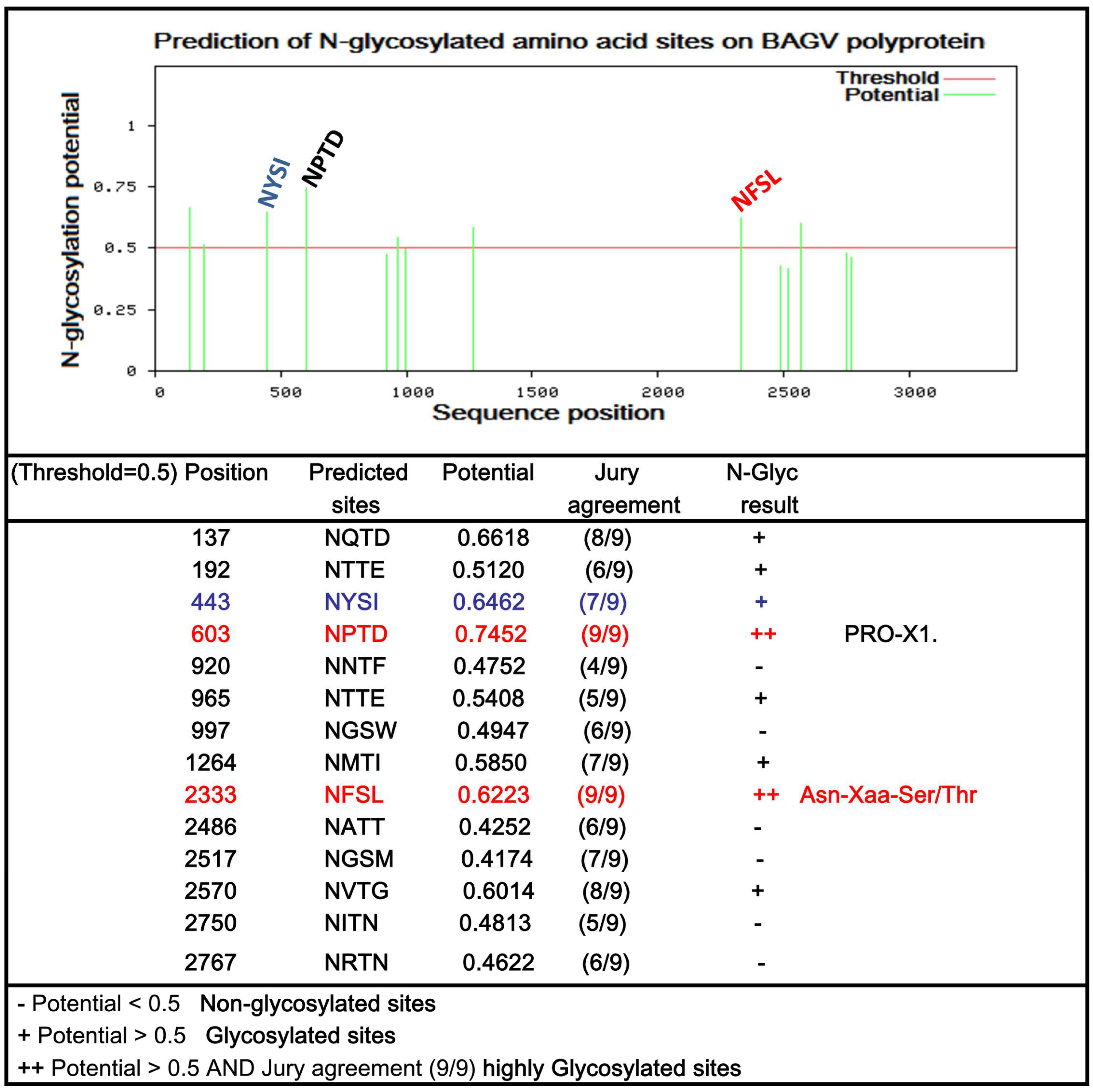
3.3. Predicted N-Glycosylated Amino Acid Sites
3.4. Predicted Structural RNA Domains on UTRs of BAGV Genome
3.5. Maximum Likelihood Tree
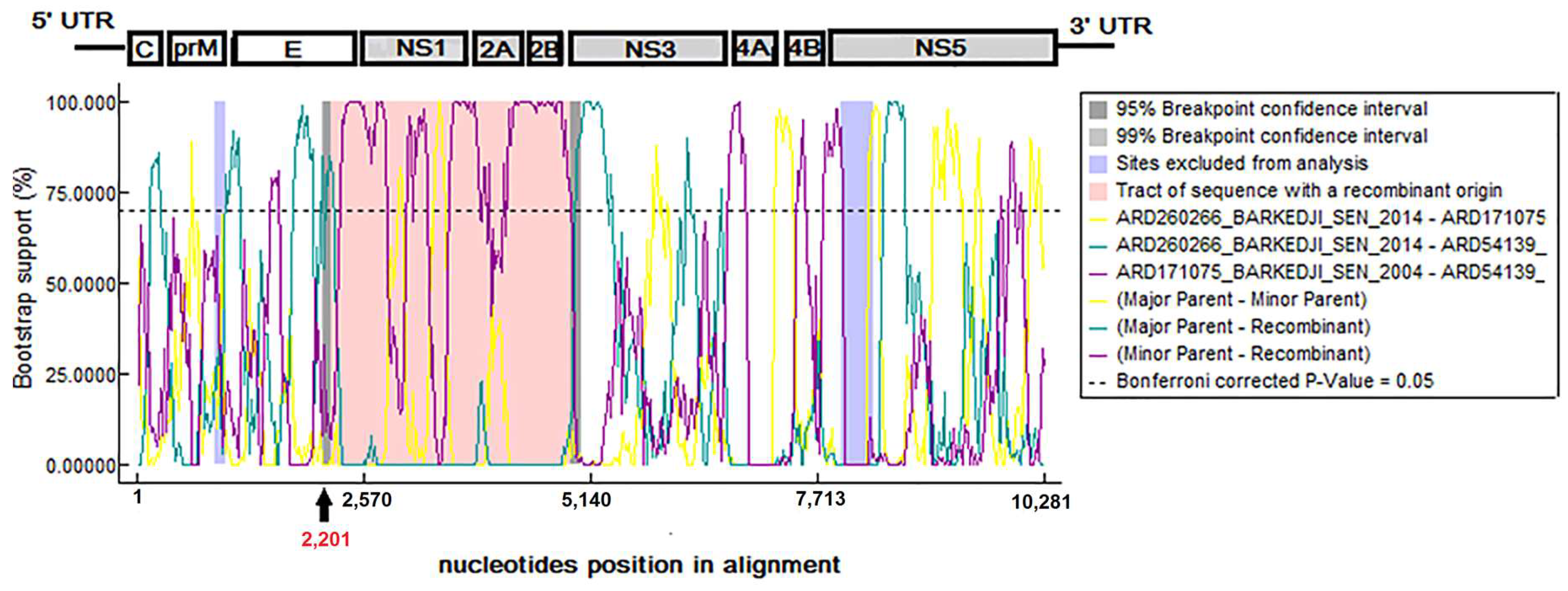
3.6. Evidence of Recombination Events
3.7. Positive Selection Pressures
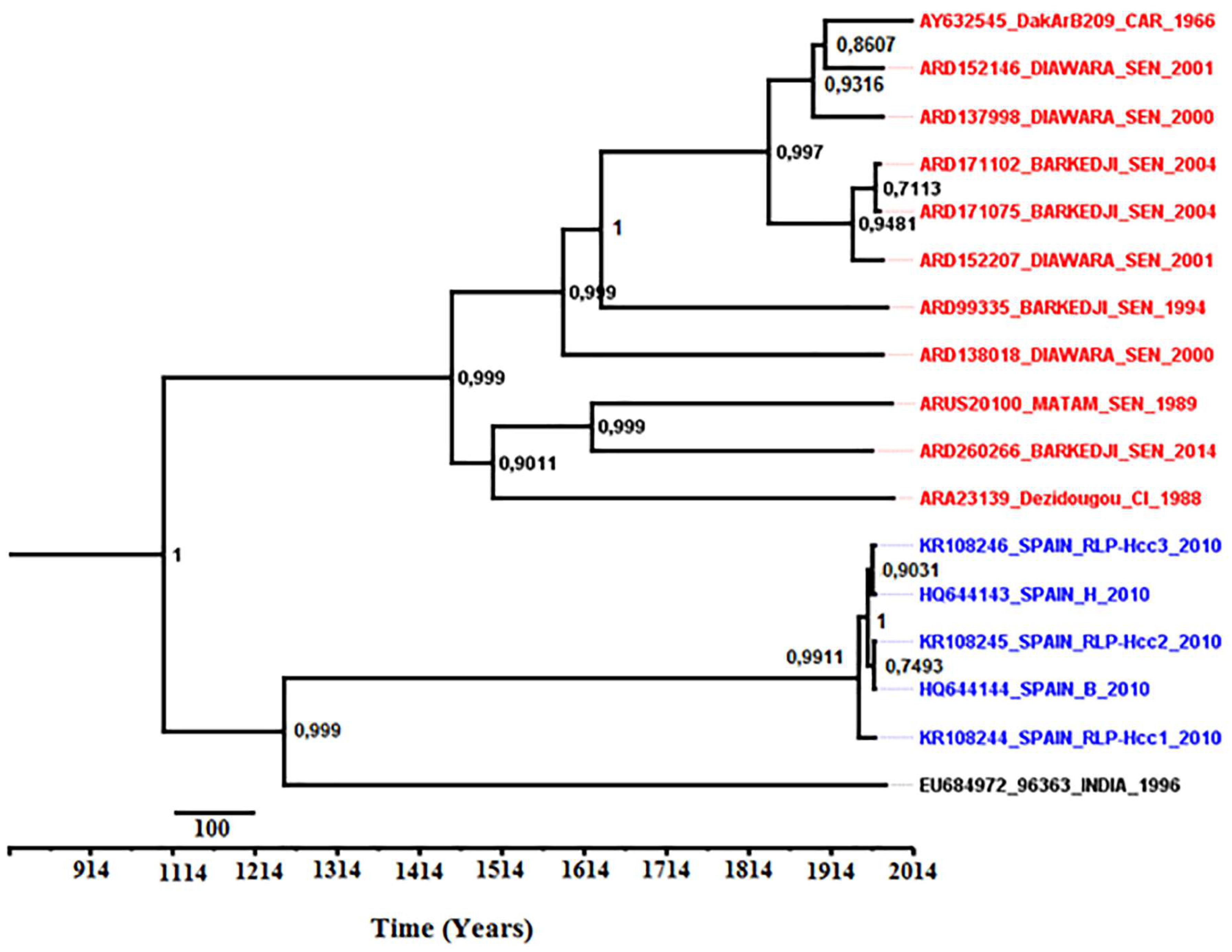
3.8. Phylodynamics of Bagaza Virus
3.9. Codon Adaptation Indexes of Viral Coding Genes
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Digoutte, J.P. Bagaza (BAG) strain: Dak Ar B 209. Am. J. Trop. Med. Hyg. 1978, 27, 376–3777. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.J.; Rice, C.M. Flaviviridae. In Fields Virology, 16th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and W: Philadelphia, PA, USA, 2013; pp. 712–746. [Google Scholar]
- Kuno, G.; Chang, G.J. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Diallo, M.; Nabeth, P.; Ba, K.; Sall, A.A.; Ba, Y.; Mondo, M. Mosquito vectors of the 1998–1999 outbreak of Rift Valley fever and other arboviruses (Bagaza, Sanar, Wesselsbron and West Nile) in Mauritania and Senegal. Med. Vet. Entomol. 2005, 19, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Traore-Lamizana, M.; Zeller, H.G.; Mondo, M.; Hervy, J.P.; Adam, F.; Digoutte, J.P. Isolations of West Nile and Bagaza viruses from mosquitoes (Diptera: Culicidae) in central Senegal (Ferlo). J. Med. Entomol. 1994, 31, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.W.; Tammariello, R.F.; Linthicum, K.J.; Dohm, D.J.; Digoutte, J.P.; Calvo-Wilson, M.A. Arbovirus isolations from mosquitoes collected during 1988 in the Senegal River basin. Am. J. Trop. Med. Hyg. 1992, 47, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Bondre, V.P.; Sapkal, G.N.; Yergolkar, P.N.; Fulmali, P.V.; Sankararaman, V.; Ayachit, V.M.; Mishra, A.C.; Gore, M.M. Genetic characterization of Bagaza virus (BAGV) isolated in India and evidence of anti-BAGV antibodies in sera collected from encephalitis patients. J. Gen. Virol. 2009, 90, 2644–2649. [Google Scholar] [CrossRef] [PubMed]
- Sudeep, A.B.; Bondre, V.P.; Mavale, M.S.; Ghodke, Y.S.; George, R.P.; Aher, R.V.; Gokhale, M.D. Preliminary findings on Bagaza virus (Flavivirus: Flaviviridae) growth kinetics, transmission potential transovarial transmission in three species of mosquitoes. Indian J. Med. Res. 2013, 138, 257–261. [Google Scholar] [PubMed]
- Agüero, M.; Fernández-Pinero, J.; Buitrago, D.; Sánchez, A.; Elizalde, M.; San Miguel, E.; Villalba, R.; Llorente, F.; Jiménez-Clavero, M.A. Bagaza virus in partridges and pheasants, Spain, 2010. Emerg. Infect. Dis. 2011, 17, 1498–1501. [Google Scholar] [CrossRef] [PubMed]
- García-Bocanegra, I.; Zorrilla, I.; Rodríguez, E.; Rayas, E.; Camacho, L.; Redondo, I.; Gómez-Guillamón, F. Monitoring of the Bagaza virus epidemic in wild bird species in Spain, 2010. Transbound. Emerg. Dis. 2013, 60, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pinero, J.; Davidson, I.; Elizalde, M.; Perk, S.; Khinich, Y.; Jiménez-Clavero, M.A. Bagaza virus and Israel turkey meningoencephalomyelitis virus are a single virus species. J. Gen. Virol. 2014, 95, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Sudeep, A.B.; Bondre, V.; George, R.; Ghodke, Y.S.; Aher, R.V.; Gokhale, M.D. Bagaza virus inhibits Japanese encephalitis & West Nile virus replication in Culex tritaeniorhynchus & Cx. quinquefasciatus mosquitoes. Indian J. Med. Res. 2015, 142 (Suppl. 1), S44–S51. [Google Scholar] [PubMed]
- Gamino, V.; Gutiérrez-Guzmán, A.V.; Fernández-de-Mera, I.G.; Ortíz, J.A.; Durán-Martín, M.; de la Fuente, J.; Gortázar, C.; Höfle, U. Natural Bagaza virus infection in game birds in southern Spain. Vet. Res. 2012, 43, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0. Mol. Biol. Evol. 2015. submitted. [Google Scholar]
- Digoutte, J.P.; Calvo-Wilson, M.A.; Mondo, M.; Traore-Lamizana, M.; Adam, F. Continuous cell lines and immune ascitic fluid pools in arbovirus detection. Res. Virol. 1992, 143, 417–422. [Google Scholar] [CrossRef]
- Patel, P.; Landt, O.; Kaiser, M.; Faye, O.; Koppe, T.; Lass, U.; Sall, A.A.; Niedrig, M. Development of one-step quantitative reverse transcription PCR for the rapid detection of flaviviruses. Virol. J. 2013, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. The UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Maust, B.S.; Nickle, D.C.; Learn, G.H.; Liu, Y.; Heath, L.; Sergei, L.; Pond, K.; Mullins, J.I. DIVEIN: A Web Server to Analyze Phylogenies, Sequence Divergence, Diversity, and Informative Sites. Biotechniques 2010, 48, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Jung, E.; Brunak, S. Prediction of N-glycosylation sites in human proteins. Proteomics 2004, 4, 1633–1649. [Google Scholar]
- Gruber, A.R.; Neuböck, R.; Hofacker, I.L.; Washietl, S. The RNAz web server: Prediction of thermodynamically stable and evolutionarily conserved RNA structures. Nucleic Acids Res. 2007, 35, W335–W338. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [PubMed]
- Hoff, M.; Orf, S.; Riehm, B.; Darriba, D.; Stamatakis, A. Does the choice of nucleotide substitution models matter topologically? BMC Bioinform. 2016, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2: Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K. The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 2002, 54, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky, P.S.L. FUBAR: A fast, unconstrained Bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol 2013, 30, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.C.M.; Iamarino, A.; Neto, D.F.L.; Sall, A.A.; Zanotto, P.M.A. Spread of the pandemic Zika virus lineage is associated with NS1 codon usage adaptation in humans. BioRxiv 2015, 2015, 032839. [Google Scholar]
- Shin, Y.C.; Bischof, G.F.; Lauer, W.A.; Desrosiers, R.C. Importance of codon usage for the temporal regulation of viral gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 14030–14035. [Google Scholar] [CrossRef] [PubMed]
- Nasrullah, I.; Butt, A.M.; Tahir, S.; Idrees, M.; Tong, Y. Genomic analysis of codon usage shows influence of mutation pressure, natural selection, and host features on Marburg virus evolution. BMC Evol. Biol. 2015, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Puigbo, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Puigbo, P.; Bravo, I.G.; Garcia-Vallve, S. E-CAI: A novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinform. 2008, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sanlés, A.; Ríos-Marco, P.; Romero-López, C.; Berzal-Herranz, A. Functional information stored in the conserved structural RNA domains of the flavivirus genome. Front. Microbiol. 2017, 546, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Twiddy, S.S.; Holmes, E.C. The extent of homologous recombination in members of the genus Flavivirus. J. Gen. Virol. 2003, 84, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, A.; Shackelton, L.A.; Holmes, E.C. Family level phylogenies reveal modes of macroevolution in RNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell 2015, 162, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Thurner, C.; Witwer, C.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004, 85, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Bidet, K.; Garcia-Blanco, M.A. Flaviviral RNAs: Weapons and targets in the war between virus and host. Biochem. J. 2014, 462, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Yue, L.; Li, X.; Yu, X.; Zhao, H.; Jiang, Z.; Qin, E.; Qin, C. Identification and characterization of small sub-genomic RNAs in dengue 1–4 virus-infected cell cultures and tissues. Biochem. Biophys. Res. Commun. 2010, 391, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.D. Functional non-coding RNAs derived from the flavivirus 3 untranslated region. Virus Res. 2015, 206, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.; Hsu, T.W.; Chen, Y.L.; Liu, S.F.; Tsai, Y.J.; Lin, Y.T.; Chen, Y.S.; Fan, Y.H. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 2013, 166, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Polacek, C.; Friebe, P.; Harris, E. Poly (A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency. J. Gen. Virol. 2009, 90, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Villordo, S.M.; Carballeda, J.M.; Filomatori, C.V.; Gamarnik, A.V. RNA Structure Duplications and Flavivirus Host Adaptation. Trends Microbiol. 2016, 24, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; Khromykh, A.A.; Asgari, S. West Nile virus encodes a microRNA-like small RNA in the 3′ untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res. 2012, 40, 2210–2223. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, W.; Meng, G.; Zhao, K.; Gu, J.; Chen, P.; Cao, R. Isolation and genome characterization of a novel duck Tembusu virus with a 74 nucleotide insertion in the 3′ non-translated region. Avian Pathol. 2015, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Setoh, Y.X.; Hall, R.A.; Khromykh, A.A. Post-translational regulation and modifications of flavivirus structural proteins. J. Gen. Virol. 2015, 96, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Lodeiro, M.F.; Filomatori, C.V.; Gamarnik, A.V. Structural and functional studies of the promoter element for Dengue virus RNA replication. J. Virol. 2009, 83, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.A.; Manlucu, L.R.; Gilmore, L.E. A trade-off in replication in mosquito versus mammalian systems conferred by a point mutation in the NS4B protein of dengue virus type 4. Virology 2003, 312, 222–232. [Google Scholar] [CrossRef]
- Rastogi, M.; Sharma, N.; Singh, S.K. Flavivirus NS1: A multifaceted enigmatic viral protein. Virol. J. 2016, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Gebhard, L.G.; Iglesias, N.G.; Byk, L.A.; Filomatori, C.V.; de Maio, F.A.; Gamarnik, A.V. A Proline-Rich N-Terminal Region of the Dengue Virus NS3 Is Crucial for Infectious Particle Production. J. Virol. 2016, 90, 5451–5461. [Google Scholar] [CrossRef] [PubMed]
- Diallo, D.; Ba, Y.; Dia, I.; Sall, A.A.; Diallo, M. Evaluation of the efficiency of bird-baited traps for sampling potential West Nile fever mosquito vectors (Diptera: Culicidae) in Senegal. Parasite 2010, 17, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Nikolay, B.; Diallo, M.; Faye, O.; Boye, C.S.; Sall, A.A. Vector Competence of Culex neavei (Diptera: Culicidae) for Usutu Virus. Am. J. Trop. Med. Hyg. 2012, 86, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Fall, G.; Diallo, M.; Loucoubar, C.; Faye, O.; Sall, A.A. Vector Competence of Culex neavei and Culex quinquefasciatus (Diptera: Culicidae) from Senegal for Lineages 1, 2, Koutango and a Putative New Lineage of West Nile virus. Am. J. Trop. Med. Hyg. 2014, 90, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Zmurko, J.; Neyts, J.; Dallmeier, K. Flaviviral NS4b, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention. Rev. Med. Virol. 2015, 25, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Fischl, W.; Scaturro, P.; Cortese, M.; Kallis, S.; Bartenschlager, M.; Fischer, B.; Bartenschlager, R. A Combined Genetic-Proteomic Approach Identifies Residues within Dengue Virus NS4B Critical for Interaction with NS3 and Viral Replication. J. Virol. 2015, 89, 7170–7186. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.; Li, T.; McCune, B.T. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J. Virol. 2012, 86, 7360–7371. [Google Scholar] [CrossRef] [PubMed]
- Naik, N.G.; Wu, H.N. Mutation of Putative N-Glycosylation Sites on Dengue Virus NS4B Decreases RNA Replication. J. Virol. 2015, 89, 6746–6760. [Google Scholar] [CrossRef] [PubMed]
- Fall, G.; Di Paola, N.; Faye, M. Biological and phylogenetic characteristics of West African lineages of West Nile virus. PLoS Negl. Trop. Dis. 2017, 11, e0006078. [Google Scholar] [CrossRef] [PubMed]
- Hacker, K.; White, L.; de Silva, A.M. N-linked glycans on dengue viruses grown in mammalian and insect cells. J. Gen. Virol. 2009, 90, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Alen, M.M.; Dallmeier, K.; Balzarini, J.; Neyts, J.; Schols, D. Crucial role of the N-glycans on the viral E-envelope glycoprotein in DC-SIGN-mediated dengue virus infection. Antivir. Res. 2012, 96, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.L.; Pierson, T.C.; Sanchez, M.D.; Ahmed, A.A.; Murtadha, M.M. N-linked glycosylation of west nile virus envelope proteins influences particle assembly and infectivity. J. Virol. 2005, 79, 13262–13274. [Google Scholar] [CrossRef] [PubMed]
- Faye, O.; Freire, C.C.; Iamarino, A.; Faye, O.; de Oliveira, J.V.; Diallo, M.; Zanotto, P.M.; Sall, A.A. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl. Trop. Dis. 2014, 8, e2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doores, K.J. The HIV glycan shield as a target for broadly neutralizing antibodies. FEBS J. 2015, 282, 4679–4691. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Tortorici, M.A.; Frenz, B.; Snijder, J.; Li, W.; Rey, F.A. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 2016, 23, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yu, H.; Dong, Y.; Yang, J.; Ye, W.; Wang, Y. Characterization of N-Glycan Structures on the Surface of Mature Dengue 2 Virus Derived from Insect Cells. PLoS ONE 2015, 10, e0132122. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Y.; Wang, S.; Zhang, Y.; Zu, X.; Zhou, Z.; Zhang, B.; Xiao, G. Structure-based mutational analysis of several sites in the E protein: Implications for understanding the entry mechanism of Japanese encephalitis virus. J. Virol. 2015, 89, 5668–5686. [Google Scholar] [CrossRef] [PubMed]
- Szentpáli-Gavallér, K.; Lim, S.M.; Dencső, L.; Bányai, K.; Koraka, P.; Osterhaus, A.D.; Bálint, Á. In vitro and in vivo evaluation of mutations in the NS region of Lineage 2 West Nile virus associated with neuroinvasiveness in a mammalian model. Viruses 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Waman, V.P.; Kolekar, P.; Ramtirthkar, M.R.; Kale, M.M.; Kulkarni-Kale, U. Analysis of genotype diversity and evolution of Dengue virus serotype 2 using complete genomes. PeerJ 2016, 4, e2326. [Google Scholar] [CrossRef] [PubMed]
- Martynova, E.U.; Schal, C.; Mukha, D.V. Effects of recombination on densovirus phylogeny. Arch. Virol. 2016, 161, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Suzuki, Y.; Gojobori, T. A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol. Biol. Evol. 2004, 21, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.; Daly, J.M.; Nisalak, A.; Solomon, T. Recombination and positive selection identified in complete genome sequences of Japanese encephalitis virus. Arch. Virol. 2012, 157, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Roehrig, J.T. Antigenic structure of flavivirus proteins. Adv. Virus Res. 2003, 59, 141–175. [Google Scholar] [PubMed]
- Bennett, S.N.; Holmes, E.C.; Chirivella, M.; Rodriguez, D.M.; Beltran, M.; Vorndam, V. Molecular evolution of dengue 2 virus in Puerto Rico: Positive selection in the viral envelope accompanies clade reintroduction. J. Gen. Virol. 2006, 87, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Chwan-Chuen, K.; Day-Yu, C.; Li-Jung, C.; Gwong-Jen, J.C.; Ting-Hsiang, L.; Yin-Chang, W.; Jyh-Hsiung, H. Comparative analysis of full genomic sequences among different genotypes of dengue virus type 3. Virol. J. 2008, 5, 63. [Google Scholar]
- Sironi, M.; Forni, D.; Clerici, M.; Cagliani, R. Nonstructural Proteins Are Preferential Positive Selection Targets in Zika Virus and Related Flaviviruses. PLoS Negl. Trop. Dis. 2016, 10, e0004978. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular localization and membrane topology of the Dengue virus type 2 Non-structural protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar] [CrossRef] [PubMed]
- Maringer, K.; Fernandez-Sesma, A. Message in a bottle: Lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev. 2014, 25, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Adams, J.; Chen, P.; Guo, Z.Y.; Zhong, X.F.; Fang, W. Comparison of genotypes I and III in Japanese encephalitis virus reveals distinct differences in their genetic and host diversity. J. Virol. 2014, 88, 11469–11479. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Xu, T.; Hunke, C.; Gruber, G.; Vasudevan, S.G. Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 2008, 82, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.P.; Decroly, E.; Malet, H.; Selisko, B.; Benarroch, D. Structural and functional analysis of methylation and 5′-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J. Mol. Biol. 2007, 372, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Añez, G.; Grinev, A.; Chancey, C.; Ball, C.; Akolkar, N.; Land, K.J.; Winkelman, V.; Stramer, S.L.; Kramer, L.D.; Rios, M. Evolutionary dynamics of West Nile virus in the United States, 1999–2011: Phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl. Trop. Dis. 2013, 7, e2245. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.U.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife 2015, 4, e08347. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, N.; Freire, C.C.M.; Zanotto, P.M.A. Does adaptation to vertebrate codon usage relate to flavivirus emergence potential? PLoS ONE 2018, 13, e0191652. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome wide patterns: With emphasis on insect genomes. Biol. Rev. 2013, 88, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K.; Severson, D.W. Bicluster pattern of codon context usages between flavivirus and vector mosquito Aedes aegypti: Relevance to infection and transcriptional response of mosquito genes. Mol. Genet. Genom. 2014, 289, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Forrester, N.; Tsetsarkin, K.; Vasilakis, N.; Weaver, S.C. Factors shaping the adaptive landscape for arborviruses: Implications for the emergence of disease. Future Microbiol. 2013, 8, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Vasilakis, N.; Brault, A.C.; Powers, A.M.; Tripet, F.; Weaver, S.C. Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl. Acad. Sci. USA 2008, 105, 6970–6975. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.P.; Travassos da Rosa, A.P.; Nunes, M.R.; Xiao, S.Y.; Tesh, R.B. Pathogenesis of Modoc virus (Flaviviridae; Flavivirus) in persistently infected hamsters. Am. J. Trop. Med. Hyg. 2013, 88, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Constantine, D.G.; Woodall, D.F. Latent Infection of Rio Bravo Virus in Salivary Glands of Bats. Public Health Rep. 1964, 79, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Blitvich, B.; Firth, A. Insect-Specific Flaviviruses: A Systematic Review of Their Discovery, Host Range, Mode of Transmission, Superinfection Exclusion Potential and Genomic Organization. Viruses 2015, 7, 1927–1959. [Google Scholar] [CrossRef] [PubMed]
- Beasley, D.W.C.; McAuley, A.J.; Bente, D.A. Yellow fever virus: Genetic and phenotypic diversity and implications for detection, prevention and therapy. Antivir. Res 2015, 115, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Belcaid, M.; Nerurkar, V.R. Identification of host genes leading to West Nile virus encephalitis in mice brain using RNA-SEQ analysis. Sci. Rep. 2016, 6, 26350. [Google Scholar] [CrossRef] [PubMed]
- Ramos da Silva, S.; Gao, S.J. Zika virus: An update on epidemiology, pathology, molecular biology, and animal model. J. Med. Virol. 2016, 88, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.L.; Dimitrov, K.M.; Afonso, C.L. Genome-wide analysis reveals class and gene specific codon usage adaptation in avian paramyxoviruses 1. Infect. Genet. Evol. 2017, 58, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Canfield, S.G.; Stebbins, M.J.; Morales, B.S.; Asai, S.W.; Vatine, G.D.; Svendsen, C.N.; Palecek, S.P.; Shusta, E.V. An Isogenic Blood-Brain Barrier Model Comprising Brain Endothelial Cells, Astrocytes and Neurons Derived from Human Induced Pluripotent Stem Cells. J. Neurochem. 2016, 140, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shin, S.; Jha, B.S.; Liu, Q.; Sheng, J.; Li, F.; Zhan, M.; Davis, J.; Bharti, K.; Zeng, X.; et al. Efficient and rapid derivation of primitive neural stem cells and generation of brain subtype neurons from human pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 862–870. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence 5′-3′ | Direction | Position on Genome | Melting Temperature (°C) |
|---|---|---|---|---|
| 5′raceBAGV1 | CATCAATCCGACATCCAGAG | Antisense | Envelope | 53 |
| 5′raceBAGV2 | CCTTTCGGAAGCTTTTCAAG | Antisense | Envelope | 53 |
| BAG28F | TTGACAGCTCAACACAAGTGC | Sense | Envelope | 55 |
| BAG1037R | CCATCACGACATCAATCCAC | Antisense | Envelope | 55 |
| BAG572F | GCTCTGGATGTCGGATTGAT | Sense | Envelope | 55 |
| BAG2069R | TTGTCCCCGATGATGATGTA | Antisense | NS1 | 55 |
| BAG3SEG1F | TCATTTCGAGTTGGCTGTGT | Sense | NS1 | 55 |
| BAG3SEG1R | TATTGGACATGGGTGGAGTG | Antisense | NS2B | 55 |
| BAG3SEG2F | GTGTAAGGTCCGTGGGAAGA | Sense | NS2A | 55 |
| BAG3SEG2R | CAAACCAATCAGCACTCCAC | Antisense | NS3 | 55 |
| BAG3538F | GAACCATTTCAGCTGGGTGT | Sense | NS3 | 55 |
| BAG5064R | CCGACAAGAATGCCATTACC | Antisense | NS4A | 55 |
| BAG4825F | TCGTATGGAGGACCTTGGAA | Sense | NS3 | 55 |
| BAG6324R | CCAAAGCTCAACTGGGTTGT | Antisense | NS4B | 55 |
| BAG6SEG1F | CGAGCCGGGTTATTGATAGT | Sense | NS4B | 55 |
| BAG6SEG1R | ACCTGCTGCTGTTCTCCTTT | Antisense | NS5 | 55 |
| BAG6SEG2F | GACGTTTTTGACACCACTGC | Sense | NS5 | 55 |
| BAG6SEG2R | GACGCGGTCTTCTACCATTT | Antisense | NS5 | 55 |
| BAG8234F | GAAAGAACTGGAACGGATGC | Sense | NS5 | 55 |
| BAG9750R | CTCGGGGATGTCTTTTCTGA | Antisense | NS5 | 55 |
| BAG9329F | AGAATGGACCCAGAGCACAG | Sense | NS5 | 55 |
| BAG10853R | TCCCAGGTGTCAATATGCTG | Antisense | NS5 | 55 |
| 3′raceBAGV1 | AAAGCGCTCAATACCGACTC | Sense | NS5 | 53 |
| 3′raceBAGV2 | AGTCAGGCCACAGGTTTTGT | Sense | NS5 | 53 |
| Isolate | Origin | Year of Isolation | Specie | Accession Numbers |
|---|---|---|---|---|
| ArA23139 | Dezidougou (CI) | 1988 | Culex poicilipes | MF380429 |
| ArD54139 | Dakar-Bango (SEN) | 1989 | Culex poicilipes | MF380430 |
| ArUS20100 | Matam (SEN) | 1989 | Culex poicilipes | MF380424 |
| ArD99335 | Barkedji (SEN) | 1994 | Culex neavei | MF380431 |
| ArD137998 | Diawara (SEN) | 2000 | Culex poicilipes | MF380425 |
| ArD138018 | Diawara (SEN) | 2000 | Culex poicilipes | MF380426 |
| ArD152146 | Diawara (SEN) | 2001 | Culex poicilipes | MF380433 |
| ArD152207 | Diawara (SEN) | 2001 | Culex poicilipes | MF380432 |
| ArD171075 | Barkedji (SEN) | 2004 | Culex poicilipes | MF380427 |
| ArD171102 | Barkedji (SEN) | 2004 | Culex poicilipes | MF380428 |
| ArD260266 | Barkedji (SEN) | 2014 | Culex neavei | MF380434 |
| Genomic Regions | AY632545_DakAR B209_CAR_1966 | HQ644143_Spain_H_2010 | Isolates Sequenced in This Study |
|---|---|---|---|
| 5′ UTR | 94 nt | 94 nt | 94 nt |
| Capsid | 122 aa | 122 aa | 122 aa |
| prM | 177 aa | 177 aa | 177 aa |
| Envelope | 501 aa | 501 aa | 501 aa |
| NS1 | 342 aa | 342 aa | 342 aa |
| NS2A | 226 aa | 226 aa | 226 aa |
| NS2B | 132 aa | 132 aa | 132 aa |
| NS3 | 619 aa | 619 aa | 619 aa |
| NS4A | 126 aa | 126 aa | 126 aa |
| 2K | 23 aa | 23 aa | 23 aa |
| NS4B | 253 aa | 253 aa | 253 aa |
| NS5 | 905 aa | 905aa | 905 aa |
| 3′ UTR | 566 nt | 439 nt | 576 nt |
| Total length | 10,941 nt | 10,794 nt | 10,951 nt |
| Gene | Amino Acid Motifs Previously Described in MBFVs # | Cons * on Used NKVFs/ISFs Genome | Positions on BAGV Genome | Cons * on BAGV | Except on These BAGV Isolates | Replaced by This Consensus Sequence |
|---|---|---|---|---|---|---|
| E | DRGWGNGC | YES | 387–394 | YES | ARD171075_BARKEDJI_SEN_2004 | ARSRGNGC |
| GLFGKGS | only on NKVFs | 395–401 | YES | ARD171102_BARKEDJI_SEN_2004 | GLFAKGS | |
| GHLKCRV | NO RBV (GHVDCRV) ModV (GHVSCKV) | 573–579 | YES | |||
| PFGDSYIV | NO | 667–675 | NO | All BAGV isolates | PFGDSFILV | |
| NS1 | DTAWDFGS | NO | 712–719 | YES | ARD171102_BARKEDJI_SEN_2004 | DPAWDFGS |
| EU684972_96363_India_1996 | DAAWDFGS | |||||
| GCWYGMEI | only on NKVFs | 1118–1125 | YES | |||
| YGMEIRP | YES | 1120–1127 | YES | ARD54139_DAKAR-BANGO_SEN_1989 | YGMEIRT | |
| NS3 | GTSGSPI | YES | 1633–1639 | YES | ||
| GLYGNG | only on NKVFs and CxFV | 1648–1653 | YES | ARUS20100_MATAM_SEN_1989 | GVYGNG | |
| LAPTRVV | YES | 1722–1728 | YES | ARD137998_DIAWARA_SEN_2000 | LPLTRLV | |
| ARD138018_DIAWARA_SEN_2000 | SPLTRLV | |||||
| ARD171075_BARKEDJI_SEN_2004 | SAPTRLV | |||||
| DVMCHATF | Only on NKVFs | 1759–1766 | NO | ARD138018_DIAWARA_SEN_2000 | DVMCHAPL | |
| Other BAGV isolates | DVMCHATL | |||||
| MDEAHF | YES | 1784–1789 | YES | ARD138018_DIAWARA_SEN_2000 | MYEAHF | |
| SIAARGY | YES | 1794–1800 | YES | |||
| MTATPPG | YES | 1815–1821 | YES | |||
| ISEMGAN | YES | 1911–1917 | YES | |||
| SAAQRRGR | YES | 1954–1961 | YES | |||
| NS5 | DLGCGRG | YES | 2601–2607 | YES | ||
| SRNSTHEMY | YES | 2734–2741 | NO | ARD54139_DAKAR-BANGO_SEN_1989 | WRNPNHEMY | |
| Other BAGV isolates | SRNSNHEMY | |||||
| NMMGKREKK | YES | 2977–2986 | YES | |||
| ADDTAGWDT | YES | 3056–3064 | YES | |||
| WMTTEDML | YES | 3330–3337 | YES |
| Proteins | Number of Sites Detected by Method | Evidence of Positive Selection | ||||
|---|---|---|---|---|---|---|
| SLAC (p < 0.1) | FUBAR (Posterior Probability ≥ 0.9) | MEME (p < 0.1) | Branch-Site REL (p < 0.05) | |||
| Capsid | Sites under negative selection (dN/dS < 1) | 0 | 4 | 0 | 0 | YES |
| Sites under positive selection (dN/dS > 1) | 0 | 2 | 2 | 0 | ||
| prM | Sites under negative selection (dN/dS < 1) | 6 | 11 | 0 | 0 | NO |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 0 | 0 | ||
| E | Sites under negative selection (dN/dS < 1) | 15 | 88 | 0 | 0 | YES |
| Sites under positive selection (dN/dS > 1) | 0 | 1 | 15 | 4 | ||
| NS1 | Sites under negative selection (dN/dS < 1) | 7 | 33 | 0 | 0 | YES |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 11 | 3 | ||
| NS2A | Sites under negative selection (dN/dS < 1) | 3 | 9 | 0 | 0 | YES |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 1 | 1 | ||
| NS2B | Sites under negative selection (dN/dS < 1) | 4 | 8 | 0 | 0 | NO |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 0 | 0 | ||
| NS3 | Sites under negative selection (dN/dS < 1) | 17 | 63 | 0 | 0 | YES |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 4 | 1 | ||
| NS4A | Sites under negative selection (dN/dS < 1) | 2 | 5 | 0 | 0 | NO |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 0 | 0 | ||
| NS4B | Sites under negative selection (dN/dS < 1) | 2 | 14 | 0 | 0 | YES |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 3 | 1 | ||
| NS5 | Sites under negative selection (dN/dS < 1) | 0 | 274 | 0 | 0 | YES |
| Sites under positive selection (dN/dS > 1) | 0 | 0 | 10 | 1 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faye, M.; Faye, O.; Diagne, M.M.; Fall, G.; Weidmann, M.; Sembene, M.; Sall, A.A.; Faye, O. Full-Genome Characterization and Genetic Evolution of West African Isolates of Bagaza Virus. Viruses 2018, 10, 193. https://doi.org/10.3390/v10040193
Faye M, Faye O, Diagne MM, Fall G, Weidmann M, Sembene M, Sall AA, Faye O. Full-Genome Characterization and Genetic Evolution of West African Isolates of Bagaza Virus. Viruses. 2018; 10(4):193. https://doi.org/10.3390/v10040193
Chicago/Turabian StyleFaye, Martin, Oumar Faye, Moussa Moise Diagne, Gamou Fall, Manfred Weidmann, Mbacke Sembene, Amadou Alpha Sall, and Ousmane Faye. 2018. "Full-Genome Characterization and Genetic Evolution of West African Isolates of Bagaza Virus" Viruses 10, no. 4: 193. https://doi.org/10.3390/v10040193
APA StyleFaye, M., Faye, O., Diagne, M. M., Fall, G., Weidmann, M., Sembene, M., Sall, A. A., & Faye, O. (2018). Full-Genome Characterization and Genetic Evolution of West African Isolates of Bagaza Virus. Viruses, 10(4), 193. https://doi.org/10.3390/v10040193