Interferon Independent Non-Canonical STAT Activation and Virus Induced Inflammation


{kind=link}
Abstract
:1. Introduction
2. Canonical and Non-Canonical JAK/STAT Activation
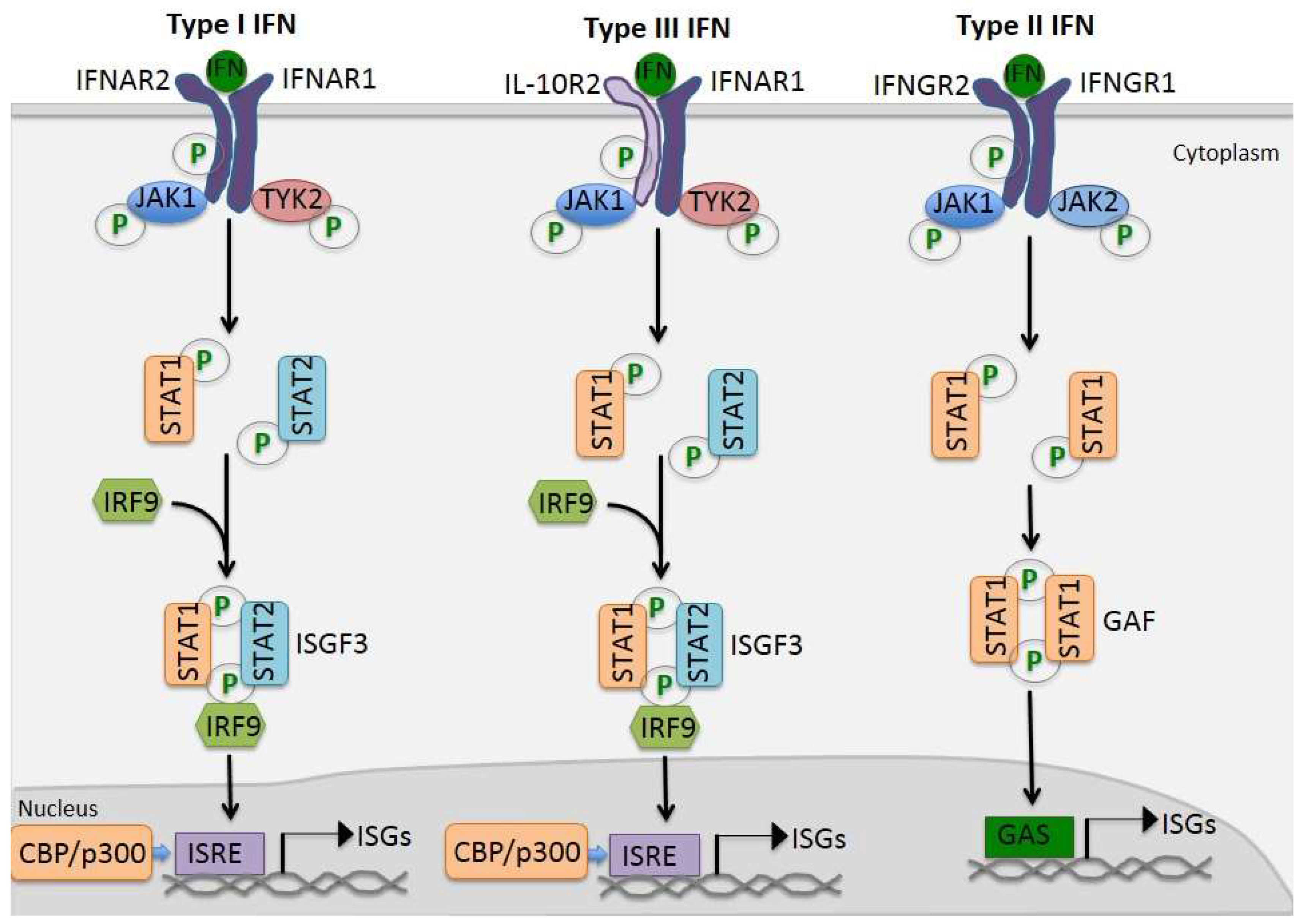
2.1. Tyrosine Phosphorylation-Dependent Canonical JAK/STAT Activation
2.2. Tyrosine Unphosphorylated STATs during Non-Canonical STATs Activation
2.3. Serine Monophosphorylation of STATs during Non-Canonical STAT Activation
3. Function of STAT Family Members and Regulation of STAT Activation
3.1. Function of STAT Family Members
3.2. Regulation of the JAK/STAT Pathway
3.3. Virus-Induced Serine Monophosphorylation of STATs and Inflammatory Responses during Virus Infection
4. Conclusions and Perspectives
Acknowledgments
Author contributions
Conflicts of Interest
References
- Gonzalez-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Nan, G.; Zhang, Y.J. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 2014, 6, 4999–5027. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.; Ozmen, L.; Novelli, F.; Geuna, M.; Palestro, G.; Forni, G.; Garotta, G. Distribution of interferon-γ receptor in human tissues. Eur. J. Immunol. 1992, 22, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Lew, D.J.; Cheng, Y.S.; Levy, D.E.; Darnell, J.E., Jr. Interactions of α- and γ-interferon in the transcriptional regulation of the gene encoding a guanylate-binding protein. EMBO J. 1989, 8, 2009–2014. [Google Scholar] [PubMed]
- Lew, D.J.; Decker, T.; Darnell, J.E., Jr. α interferon and γ interferon stimulate transcription of a single gene through different signal transduction pathways. Mol. Cell. Biol. 1989, 9, 5404–5411. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. Interferons and viral infections. Biofactors 2009, 35, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Shuai, K.; Prezioso, V.R.; Darnell, J.E., Jr. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 1992, 257, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Katze, M.G.; He, Y.; Gale, M., Jr. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of interferon-stimulated genes with antiretroviral activity. Cell Host Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. STATs: Transcriptional control and biological impact. Nat. Rev. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wu, C.; Zhang, Y.J. Interplay between Janus kinase/signal transducer and activator of transcription signaling activated by type I interferons and viral antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Bente, D.A.; Alimonti, J.B.; Shieh, W.J.; Camus, G.; Stroher, U.; Zaki, S.; Jones, S.M. Pathogenesis and immune response of Crimean-Congo hemorrhagic fever virus in a STAT-1 knockout mouse model. J. Virol. 2010, 84, 11089–11100. [Google Scholar] [CrossRef] [PubMed]
- Yun, N.E.; Seregin, A.V.; Walker, D.H.; Popov, V.L.; Walker, A.G.; Smith, J.N.; Miller, M.; de la Torre, J.C.; Smith, J.K.; Borisevich, V.; et al. Mice lacking functional STAT1 are highly susceptible to lethal infection with Lassa virus. J. Virol. 2013, 87, 10908–10911. [Google Scholar] [CrossRef] [PubMed]
- Bradfute, S.B.; Stuthman, K.S.; Shurtleff, A.C.; Bavari, S. A STAT-1 knockout mouse model for Machupo virus pathogenesis. Virol. J. 2011, 8, 300. [Google Scholar] [CrossRef] [PubMed]
- Ortego, J.; de la Poza, F.; Marin-Lopez, A. Interferon α/β receptor knockout mice as a model to study bluetongue virus infection. Virus Res. 2014, 182, 35–42. [Google Scholar] [CrossRef] [PubMed]
- O'Shea, J.J. Jaks, STATs, cytokine signal transduction, and immunoregulation: Are we there yet? Immunity 1997, 7, 1–11. [Google Scholar] [CrossRef]
- Saharinen, P.; Silvennoinen, O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J. Biol. Chem. 2002, 277, 47954–47963. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Ultsch, M.; Wallweber, H.; Bir Kohli, P.; Johnson, A.R.; Eigenbrot, C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 8025–8030. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, F.Z.; Farrar, J.D. STAT2: A shape-shifting anti-viral super STAT. Jak-Stat 2013, 2, e23633. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.S.; Sanders, L.K.; Nathans, D. Cooperative transcriptional activity of Jun and STAT3 β, a short form of STAT3. Proc. Natl. Acad. Sci. USA 1995, 92, 9097–9101. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Tweardy, D.J. Granulocyte colony-stimulating factor activates a 72-kDa isoform of STAT3 in human neutrophils. J. Leukoc. Biol. 1998, 64, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; de Groot, R.P. STAT3β, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar] [CrossRef] [PubMed]
- O'Malley, J.T.; Eri, R.D.; Stritesky, G.L.; Mathur, A.N.; Chang, H.C.; Hogenesch, H.; Srinivasan, M.; Kaplan, M.H. STAT4 isoforms differentially regulate Th1 cytokine production and the severity of inflammatory bowel disease. J. Immunol. 2008, 181, 5062–5070. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.A.; Secor, V.H.; Brown, M.A. IL-4 preferentially activates a novel STAT6 isoform in mast cells. J. Immunol. 1999, 162, 2703–2708. [Google Scholar] [PubMed]
- Moriggl, R.; Gouilleux-Gruart, V.; Jahne, R.; Berchtold, S.; Gartmann, C.; Liu, X.; Hennighausen, L.; Sotiropoulos, A.; Groner, B.; Gouilleux, F. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol. Cell. Biol. 1996, 16, 5691–5700. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.; Lee, C.; Strehlow, I.; Schindler, C. Functionally distinct isoforms of STAT5 are generated by protein processing. Immunity 1997, 6, 691–701. [Google Scholar] [CrossRef]
- Steen, H.C.; Gamero, A.M. STAT2 phosphorylation and signaling. Jak-Stat 2013, 2, e25790. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000, 25, 496–502. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, T.; Imamoto, N.; Nakajima, K.; Hirano, T.; Yoneda, Y. Extracellular signal-dependent nuclear import of STAT1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997, 16, 7067–7077. [Google Scholar] [CrossRef] [PubMed]
- Nardozzi, J.; Wenta, N.; Yasuhara, N.; Vinkemeier, U.; Cingolani, G. Molecular basis for the recognition of phosphorylated STAT1 by importin α5. J. Mol. Biol. 2010, 402, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dolken, L.; Strobl, B.; Muller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008, 18, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.; Reich, N.C. Constitutive nuclear import of latent and activated STAT5a by its coiled coil domain. FASEB J 2008, 22, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Stark, G.R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef] [PubMed]
- Testoni, B.; Vollenkle, C.; Guerrieri, F.; Gerbal-Chaloin, S.; Blandino, G.; Levrero, M. Chromatin dynamics of gene activation and repression in response to interferon α (IFNα) reveal new roles for phosphorylated and unphosphorylated forms of the transcription factor STAT2. J. Biol. Chem. 2011, 286, 20217–20227. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Yang, Y.M.; Liang, F.X.; Gough, D.J.; Levy, D.E.; Sehgal, P.B. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am. J. Physiol. Cell Physiol. 2012, 302, C804–C820. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Flores Santa Cruz, D.; Sexl, V.; Gottgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016, 35, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R.; Fan, M.; Du, Z.; Yang, C.H.; Pfeffer, L.M. Unphosphorylated STAT3 regulates the antiproliferative, antiviral, and gene-inducing actions of type I interferons. Biochem. Biophys. Res. Commun. 2017, 490, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Decker, T. Emancipation from transcriptional latency: Unphosphorylated STAT5 as guardian of hematopoietic differentiation. EMBO J. 2016, 35, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhang, L.; Luo, J.; Rajasekaran, A.; Hazra, S.; Cacalano, N.; Dubinett, S.M. Unphosphorylated STAT6 contributes to constitutive cyclooxygenase-2 expression in human non-small cell lung cancer. Oncogene 2007, 26, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Nan, Y.; Shen, M.; Ritthipichai, K.; Zhu, X.; Zhang, Y.J. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J. Virol. 2010, 84, 11045–11055. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.; Hauser, H. Dynamic redistribution of STAT1 protein in IFN signaling visualized by GFP fusion proteins. Eur. J. Biochem. 1999, 260, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Cao, W.; Zhou, X.; et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal. 2017, 10, eaah4248. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.S.; Cheon, H.; Cho, C.H.; Hong, S.H.; Park, D.Y.; Seo, H.I.; Park, S.H.; Yoon, S.K.; Stark, G.R.; Shin, E.C. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc. Natl. Acad. Sci. USA 2015, 112, 10443–10448. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.K.; et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem. J. 2015, 466, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 2011, 7, e1001297. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hazan-Halevy, I.; Harris, D.M.; Li, P.; Ferrajoli, A.; Faderl, S.; Keating, M.J.; Estrov, Z. STAT-3 activates NF-κB in chronic lymphocytic leukemia cells. Mol. Cancer Res. 2011, 9, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Lopes, M.H.; Bleggi-Torres, L.F.; Suzuki, S.; Cunha, I.W.; Sanematsu, P.; Martins, V.R. Nuclear unphosphorylated STAT3 correlates with a worse prognosis in human glioblastoma. Pathol. Res. Pract. 2016, 212, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; Di Giovanni, J. Non-canonical STAT3 signaling in cancer. Mol. Carcinog. 2016, 55, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, M.; Yamamoto, M.; Tanaka, H.; Ogawa, K. Serine 727 phosphorylation of STAT3: An early change in mouse hepatocarcinogenesis induced by neonatal treatment with diethylnitrosamine. Mol. Carcinog. 2014, 53, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Sadzak, I.; Schiff, M.; Gattermeier, I.; Glinitzer, R.; Sauer, I.; Saalmuller, A.; Yang, E.; Schaljo, B.; Kovarik, P. Recruitment of STAT1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 8944–8949. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Halupa, A.; Beattie, B.K.; Mason, J.M.; Zanke, B.W.; Barber, D.L. Regulation of erythropoietin-induced STAT serine phosphorylation by distinct mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 17359–17366. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Fan, M.; Xiang, G.; Wang, J.; Zhang, X.; Guo, W.; Wu, X.; Sun, Y.; Gu, Y.; Ge, H.; et al. Diptoindonesin G promotes ERK-mediated nuclear translocation of p-STAT1 (Ser727) and cell differentiation in AML cells. Cell Death Dis. 2017, 8, e2765. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Szappanos, D.; Cheon, H.; Vogl, C.; Shukla, P.; Stark, G.R.; Sexl, V.; Schreiber, R.; Schindler, C.; et al. Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1. EMBO Rep. 2016, 17, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and non-canonical aspects of JAK-STAT signaling: Lessons from interferons for cytokine responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.M.; Gotthardt, D.; Hoermann, G.; Csiszar, A.; Wirth, S.; Berger, A.; Straka, E.; Rigler, D.; Wallner, B.; Jamieson, A.M.; et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep. 2013, 4, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.M.; Gotthardt, D.; Sexl, V. STAT1-S727—The license to kill. Oncoimmunology 2014, 3, e955441. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.A.; Rahman, N.T.; Yang, D.; Lahat, G.; Lazar, A.J.; Pollock, R.E.; Lev, D.; Liu, K. Unphosphorylated STAT1 promotes sarcoma development through repressing expression of Fas and bad and conferring apoptotic resistance. Cancer Res. 2012, 72, 4724–4732. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, R.; Nan, Y.; Zhang, L.; Zhang, Y. Induction of STAT1 phosphorylation at serine 727 and expression of proinflammatory cytokines by porcine reproductive and respiratory syndrome virus. PLoS ONE 2013, 8, e61967. [Google Scholar] [CrossRef] [PubMed]
- Koehler, M.F.; Bergeron, P.; Blackwood, E.M.; Bowman, K.; Clark, K.R.; Firestein, R.; Kiefer, J.R.; Maskos, K.; McCleland, M.L.; Orren, L.; et al. Development of a potent, specific CDK8 kinase inhibitor which phenocopies CDK8/19 knockout cells. ACS Med. Chem. Lett. 2016, 7, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Dale, T.; Clarke, P.A.; Esdar, C.; Waalboer, D.; Adeniji-Popoola, O.; Ortiz-Ruiz, M.J.; Mallinger, A.; Samant, R.S.; Czodrowski, P.; Musil, D.; et al. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat. Chem. Biol. 2015, 11, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.; Choi, S.H.; Pareek, T.K.; Kim, B.G.; Letterio, J.J. Cyclin-dependent kinase 5 represses Foxp3 gene expression and Treg development through specific phosphorylation of STAT3 at Serine 727. Mol Immunol 2015, 67 Pt B, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, T.; Mikula, M.; Zylkiewicz, E.; Dreas, A.; Wiklik, K.; Golas, A.; Wojcik, K.; Masiejczyk, M.; Wrobel, A.; Dolata, I.; et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget 2017, 8, 33779–33795. [Google Scholar] [CrossRef] [PubMed]
- Uzel, G.; Sampaio, E.P.; Lawrence, M.G.; Hsu, A.P.; Hackett, M.; Dorsey, M.J.; Noel, R.J.; Verbsky, J.W.; Freeman, A.F.; Janssen, E.; et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J. Allergy Clin. Immunol. 2013, 131, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Eckner, R.; Grossman, S.; Oldread, E.; Arany, Z.; D’Andrea, A.; Livingston, D.M. Cooperation of STAT2 and p300/CBP in signalling induced by interferon-α. Nature 1996, 383, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Bluyssen, H.A.; Levy, D.E. STAT2 is a transcriptional activator that requires sequence-specific contacts provided by STAT1 and p48 for stable interaction with DNA. J. Biol. Chem. 1997, 272, 4600–4605. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Grandvaux, N. STAT2 and IRF9: Beyond ISGF3. JAK-STAT 2013, 2, e27521. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Akira, S.; Kishimoto, T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol. Rev. 1992, 127, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, R.C.; Bierie, B.; Zhao, L.; Raz, R.; Levy, D.; Hennighausen, L. Deletion of STAT3 blocks mammary gland involution and extends functional competence of the secretory epithelium in the absence of lactogenic stimuli. Endocrinology 2002, 143, 3641–3650. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.S.; Lourenco, P.C.; Tonner, E.; Flint, D.J.; Selbert, S.; Takeda, K.; Akira, S.; Clarke, A.R.; Watson, C.J. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of STAT3. Genes Dev. 1999, 13, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of STAT3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef] [PubMed]
- Kreuzaler, P.A.; Staniszewska, A.D.; Li, W.; Omidvar, N.; Kedjouar, B.; Turkson, J.; Poli, V.; Flavell, R.A.; Clarkson, R.W.; Watson, C.J. STAT3 controls lysosomal-mediated cell death in vivo. Nat. Cell. Biol. 2011, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Mahony, R.; Gargan, S.; Roberts, K.L.; Bourke, N.; Keating, S.E.; Bowie, A.G.; O’Farrelly, C.; Stevenson, N.J. A novel anti-viral role for STAT3 in IFN-α signalling responses. Cell. Mol. Life Sci. 2017, 74, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, R.; Ma, Z.; Xiao, Y.; Nan, Y.; Wang, Y.; Lin, S.; Zhang, Y.J. Porcine reproductive and respiratory syndrome virus antagonizes JAK/STAT3 Signaling via nsp5, which induces STAT3 degradation. J. Virol. 2017, 91, e02087-16. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Quelle, F.W.; Thierfelder, W.E.; Kreider, B.L.; Gilbert, D.J.; Jenkins, N.A.; Copeland, N.G.; Silvennoinen, O.; Ihle, J.N. STAT4, a novel γ interferon activation site-binding protein expressed in early myeloid differentiation. Mol. Cell. Biol. 1994, 14, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. STAT3 and STAT4: Members of the family of signal transducers and activators of transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 4806–4810. [Google Scholar] [CrossRef] [PubMed]
- Bacon, C.M.; Petricoin, E.F., 3rd; Ortaldo, J.R.; Rees, R.C.; Larner, A.C.; Johnston, J.A.; O’Shea, J.J. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 7307–7311. [Google Scholar] [CrossRef] [PubMed]
- Thieu, V.T.; Yu, Q.; Chang, H.C.; Yeh, N.; Nguyen, E.T.; Sehra, S.; Kaplan, M.H. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 2008, 29, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Rogge, L.; D’Ambrosio, D.; Biffi, M.; Penna, G.; Minetti, L.J.; Presky, D.H.; Adorini, L.; Sinigaglia, F. The role of STAT4 in species-specific regulation of Th cell development by type I IFNs. J. Immunol. 1998, 161, 6567–6574. [Google Scholar] [PubMed]
- Farrar, J.D.; Smith, J.D.; Murphy, T.L.; Murphy, K.M. Recruitment of STAT4 to the human interferon-α/β receptor requires activated STAT2. J. Biol. Chem. 2000, 275, 2693–2697. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Dighe, A.S.; Gubler, U.; Murphy, K.M. Regulation of the interleukin (IL)-12R β 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J. Exp. Med. 1997, 185, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.H.; Sun, Y.L.; Hoey, T.; Grusby, M.J. Impaired IL-12 responses and enhanced development of Th2 cells in STAT4-deficient mice. Nature 1996, 382, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, W.E.; van Deursen, J.M.; Yamamoto, K.; Tripp, R.A.; Sarawar, S.R.; Carson, R.T.; Sangster, M.Y.; Vignali, D.A.; Doherty, P.C.; Grosveld, G.C.; et al. Requirement for STAT4 in interleukin-12-mediated responses of natural killer and T cells. Nature 1996, 382, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Vahedi, G.; Sun, H.W.; Watford, W.T.; Takatori, H.; Ramos, H.L.; Takahashi, H.; Liang, J.; Gutierrez-Cruz, G.; Zang, C.; et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity 2010, 32, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Dulek, D.E.; Newcomb, D.C.; Toki, S.; Goliniewska, K.; Cephus, J.; Reiss, S.; Bates, J.T.; Crowe, J.E., Jr.; Boyd, K.L.; Moore, M.L.; et al. STAT4 deficiency fails to induce lung Th2 or Th17 immunity following primary or secondary respiratory syncytial virus (RSV) challenge but enhances the lung RSV-specific CD8+ T cell immune response to secondary challenge. J. Virol. 2014, 88, 9655–9672. [Google Scholar] [CrossRef] [PubMed]
- Wakao, H.; Gouilleux, F.; Groner, B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 1994, 13, 2182–2191. [Google Scholar] [PubMed]
- Moriggl, R.; Sexl, V.; Piekorz, R.; Topham, D.; Ihle, J.N. STAT5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity 1999, 11, 225–230. [Google Scholar] [CrossRef]
- Moriggl, R.; Topham, D.J.; Teglund, S.; Sexl, V.; McKay, C.; Wang, D.; Hoffmeyer, A.; van Deursen, J.; Sangster, M.Y.; Bunting, K.D.; et al. STAT5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity 1999, 10, 249–259. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Harir, N.; Pecquet, C.; Kerenyi, M.; Sonneck, K.; Kovacic, B.; Nyga, R.; Brevet, M.; Dhennin, I.; Gouilleux-Gruart, V.; Beug, H.; et al. Constitutive activation of STAT5 promotes its cytoplasmic localization and association with PI3-kinase in myeloid leukemias. Blood 2007, 109, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Alexander, D.R. Fusion tyrosine kinase mediated signalling pathways in the transformation of haematopoietic cells. Leukemia 2006, 20, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.; Laurence, A.; Robinson, G.W.; Bonelli, M.; Dema, B.; Afzali, B.; Shih, H.Y.; Sun, H.W.; Brooks, S.R.; Hennighausen, L.; et al. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. eLife 2016, 5, e08384. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Schindler, U.; Henzel, W.J.; Ho, T.C.; Brasseur, M.; McKnight, S.L. An interleukin-4-induced transcription factor: IL-4 Stat. Science 1994, 265, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.X.; Migone, T.S.; Tsang, M.; Friedmann, M.; Weatherbee, J.A.; Zhou, L.; Yamauchi, A.; Bloom, E.T.; Mietz, J.; John, S.; et al. The role of shared receptor motifs and common STAT proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity 1995, 2, 331–339. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. STAT6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef]
- Linehan, L.A.; Warren, W.D.; Thompson, P.A.; Grusby, M.J.; Berton, M.T. STAT6 is required for IL-4-induced germline Ig gene transcription and switch recombination. J. Immunol. 1998, 161, 302–310. [Google Scholar] [PubMed]
- Walford, H.H.; Doherty, T.A. STAT6 and lung inflammation. JAK-STAT 2013, 2, e25301. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, H.; You, F.; Sun, W.; Zhou, X.; Chen, L.; Yang, J.; Wang, Y.; Tang, H.; Guan, Y.; et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 2011, 147, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Sharrocks, A.D. PIAS proteins and transcriptional regulation--more than just SUMO E3 ligases? Genes Dev. 2006, 20, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Toh, E.A.; Kikuchi, Y. Comparative analysis of yeast PIAS-type SUMO ligases in vivo and in vitro. J. Biochem. 2003, 133, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Muller, S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc. Natl. Acad. Sci. USA 2002, 99, 2872–2877. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of STAT3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liao, J.; Rao, X.; Kushner, S.A.; Chung, C.D.; Chang, D.D.; Shuai, K. Inhibition of STAT1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. USA 1998, 95, 10626–10631. [Google Scholar] [CrossRef] [PubMed]
- Arora, T.; Liu, B.; He, H.; Kim, J.; Murphy, T.L.; Murphy, K.M.; Modlin, R.L.; Shuai, K. PIASx is a transcriptional co-repressor of signal transducer and activator of transcription 4. J. Biol. Chem. 2003, 278, 21327–21330. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gross, M.; ten Hoeve, J.; Shuai, K. A transcriptional corepressor of STAT1 with an essential LXXLL signature motif. Proc. Natl. Acad. Sci. USA 2001, 98, 3203–3207. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Fu, Y.; Shuai, K. Distinct roles of the NH2- and COOH-terminal domains of the protein inhibitor of activated signal transducer and activator of transcription (STAT) 1 (PIAS1) in cytokine-induced PIAS1-STAT1 interaction. Proc. Natl. Acad. Sci. USA 2000, 97, 5267–5272. [Google Scholar] [CrossRef] [PubMed]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jaaskelainen, T.; Palvimo, J.J. PIAS proteins: Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Horvath, C.M.; Matunis, M.J. SUMO modification of STAT1 and its role in PIAS-mediated inhibition of gene activation. J. Biol. Chem. 2003, 278, 30091–30097. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, D.; Vanhatupa, S.; Kotaja, N.; Yang, J.; Aittomaki, S.; Janne, O.A.; Palvimo, J.J.; Silvennoinen, O. PIAS proteins promote SUMO-1 conjugation to STAT1. Blood 2003, 102, 3311–3313. [Google Scholar] [CrossRef] [PubMed]
- Droescher, M.; Begitt, A.; Marg, A.; Zacharias, M.; Vinkemeier, U. Cytokine-induced paracrystals prolong the activity of signal transducers and activators of transcription (STAT) and provide a model for the regulation of protein solubility by small ubiquitin-like modifier (SUMO). J. Biol. Chem. 2011, 286, 18731–18746. [Google Scholar] [CrossRef] [PubMed]
- Kramer, O.H.; Moriggl, R. Acetylation and sumoylation control STAT5 activation antagonistically. JAK-STAT 2012, 1, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. JAK-STAT 2013, 2, e24053. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, N.J.; Murphy, J.M.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. Regulation of Janus kinases by SOCS proteins. Biochem. Soc. Trans. 2013, 41, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and Autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, N.J.; Murphy, J.M.; Liau, N.P.; Varghese, L.N.; Laktyushin, A.; Whitlock, E.L.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 2013, 20, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Kershaw, N.J.; Murphy, J.M.; Varghese, L.N.; Laktyushin, A.; Young, S.N.; Lucet, I.S.; Norton, R.S.; Nicola, N.A. Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 2012, 36, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Vlotides, G.; Sorensen, A.S.; Kopp, F.; Zitzmann, K.; Cengic, N.; Brand, S.; Zachoval, R.; Auernhammer, C.J. SOCS-1 and SOCS-3 inhibit IFN-α-induced expression of the antiviral proteins 2,5-OAS and MxA. Biochem. Biophys. Res. Commun. 2004, 320, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Piganis, R.A.; De Weerd, N.A.; Gould, J.A.; Schindler, C.W.; Mansell, A.; Nicholson, S.E.; Hertzog, P.J. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferonα receptor (IFNAR1)-associated tyrosine kinase Tyk2. J. Biol. Chem. 2011, 286, 33811–33818. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Yasukawa, H.; Suzuki, A.; Kamizono, S.; Syoda, T.; Kinjyo, I.; Sasaki, M.; Johnston, J.A.; Yoshimura, A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 1999, 4, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Honke, N.; Shaabani, N.; Zhang, D.E.; Hardt, C.; Lang, K.S. Multiple functions of USP18. Cell Death Dis. 2016, 7, e2444. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Bohmer, F.D.; Friedrich, K. Protein tyrosine phosphatases as wardens of STAT signaling. JAK-STAT 2014, 3, e28087. [Google Scholar] [CrossRef] [PubMed]
- Icardi, L.; Mori, R.; Gesellchen, V.; Eyckerman, S.; De Cauwer, L.; Verhelst, J.; Vercauteren, K.; Saelens, X.; Meuleman, P.; Leroux-Roels, G.; et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl. Acad. Sci. USA 2012, 109, 12058–12063. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell Signal. 2013, 25, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Kosan, C.; Ginter, T.; Heinzel, T.; Kramer, O.H. STAT5 acetylation: Mechanisms and consequences for immunological control and leukemogenesis. JAK-STAT 2013, 2, e26102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Mustelin, T.; David, M. Arginine methylation of STAT1 regulates its dephosphorylation by T cell protein tyrosine phosphatase. J. Biol. Chem. 2002, 277, 35787–35790. [Google Scholar] [CrossRef] [PubMed]
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. 2015, 16, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Mowen, K.A.; Tang, J.; Zhu, W.; Schurter, B.T.; Shuai, K.; Herschman, H.R.; David, M. Arginine methylation of STAT1 modulates IFNα/β-induced transcription. Cell 2001, 104, 731–741. [Google Scholar] [CrossRef]
- Chen, W.; Daines, M.O.; Hershey, G.K. Methylation of STAT6 modulates STAT6 phosphorylation, nuclear translocation, and DNA-binding activity. J. Immunol. 2004, 172, 6744–6750. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, J.; Liu, S.; Xia, M.; Zhang, X.; Han, D.; Jiang, Y.; Wang, C.; Cao, X. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 2017, 170, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, M.; Unal, H.; Willard, B.; Yang, J.; Karnik, S.S.; Stark, G.R. Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. J. Biol. Chem. 2014, 289, 30763–30771. [Google Scholar] [CrossRef] [PubMed]
- Villarroya-Beltri, C.; Guerra, S.; Sanchez-Madrid, F. ISGylation—A key to lock the cell gates for preventing the spread of threats. J. Cell Sci. 2017, 130, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, J.; Cao, X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun. 2017, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Yan, M.; Malakhov, M.P.; Yuan, Y.; Ritchie, K.J.; Kim, K.I.; Peterson, L.F.; Shuai, K.; Zhang, D.E. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003, 17, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Przanowski, P.; Loska, S.; Cysewski, D.; Dabrowski, M.; Kaminska, B. ISG’ylation increases stability of numerous proteins including STAT1, which prevents premature termination of immune response in LPS-stimulated microglia. Neurochem. Int. 2018, 112, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gogulamudi, V.R.; Peryasamy, R.; Raghavaraju, G.; Subramania, U.; Pandey, K.N. Inhibition of HDAC Enhances STAT Acetylation, Blocks NF-κB, and Suppresses the Renal Inflammation and Fibrosis in Npr1 Haplotype Male Mice. Am. J. Physiol. Renal. Physiol. 2017, 313, F781–F795. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Kaowinn, S.; Cho, I.R.; Min, D.S.; Myung, H.; Oh, S.; Kaewpiboon, C.; Kraemer, O.H.; Chung, Y.H. Hepatitis C virus core protein enhances hepatocellular carcinoma cells to be susceptible to oncolytic vesicular stomatitis virus through down-regulation of HDAC4. Biochem. Biophys. Res. Commun. 2016, 474, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, F.; Zheng, M.; Zhu, H.; Zhao, D.; Liu, W.; Chen, Z. Inhibition of STAT1 methylation is involved in the resistance of hepatitis B virus to Interferon α. Antivir. Res. 2010, 85, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, R.; Mundt, F.; Gilthorpe, J.D.; Wolfel, S.; Gekara, N.O.; Kroger, A.; Overby, A.K. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflamm. 2016, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.B. Viral Inhibition of the IFN-induced JAK/STAT signalling pathway: development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.J. Antagonizing interferon-mediated immune response by porcine reproductive and respiratory syndrome virus. BioMed. Res. Int. 2014, 2014, 315470. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.J. Antagonizing cytokine-mediated JAK-STAT signaling by porcine reproductive and respiratory syndrome virus. Vet. Microbiol. 2016, 209, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wu, C.; Gu, G.; Sun, W.; Zhang, Y.J.; Zhou, E.M. Improved vaccine against PRRSV: Current progress and future perspective. Front. Microbiol. 2017, 8, 1635. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Mo, D.; Wang, Q.; Jia, J.; Qin, L.; Yu, X.; Niu, Y.; Zhao, X.; Liu, X.; Chen, Y. Aberrant host immune response induced by highly virulent PRRSV identified by digital gene expression tag profiling. BMC Genom. 2010, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, Y.; Jiang, Y.; Li, G.; Yan, L.; Yu, H.; Tong, G. Generation of an infectious clone of HuN4-F112, an attenuated live vaccine strain of porcine reproductive and respiratory syndrome virus. Virol. J. 2011, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, W.; Zhou, E.; Wang, S.; Hu, S.; Cai, X.; Rong, F.; Wu, J.; Xu, M.; Li, L. Dynamic changes in inflammatory cytokines in pigs infected with highly pathogenic porcine reproductive and respiratory syndrome virus. Clin. Vaccine Immunol. 2010, 17 Pt 7, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- McLaren, J.; Rowe, M.; Brennan, P. Epstein-Barr virus induces a distinct form of DNA-bound STAT1 compared with that found in interferon-stimulated B lymphocytes. J. Gen. Virol. 2007, 88, 1876–1886. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Yang, B.; Gendelman, H.E.; Persidsky, Y.; Kanmogne, G.D. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008, 111, 2062–2072. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, V.; Navarro, L.; Sample, C.E.; David, M.; Sung, S.; Swaminathan, S. The Epstein-Barr virus SM protein induces STAT1 and interferon-stimulated gene expression. J. Virol. 2003, 77, 3690–3701. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.T.; Lin, C.W. EBV-encoded miR-BART20-5p and miR-BART8 inhibit the IFN-γ-STAT1 pathway associated with disease progression in nasal NK-cell lymphoma. Am. J. Pathol. 2014, 184, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Dawson, C.W.; Takada, K.; Curnow, J.; Moody, C.A.; Sixbey, J.W.; Young, L.S. Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-κB transcription factor pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 15730–15735. [Google Scholar] [CrossRef] [PubMed]
- Weber-Nordt, R.M.; Egen, C.; Wehinger, J.; Ludwig, W.; Gouilleux-Gruart, V.; Mertelsmann, R.; Finke, J. Constitutive activation of STAT proteins in primary lymphoid and myeloid leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood 1996, 88, 809–816. [Google Scholar] [PubMed]
- Michaud, F.; Coulombe, F.; Gaudreault, E.; Paquet-Bouchard, C.; Rola-Pleszczynski, M.; Gosselin, J. Epstein-Barr virus interferes with the amplification of IFNα secretion by activating suppressor of cytokine signaling 3 in primary human monocytes. PLoS ONE 2010, 5, e11908. [Google Scholar] [CrossRef] [PubMed]
- Hooykaas, M.J.G.; van Gent, M.; Soppe, J.A.; Kruse, E.; Boer, I.G.J.; van Leenen, D.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Ressing, M.E.; Wiertz, E.; et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. J. Immunol. 2017, 198, 4062–4073. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, L.N.; Qin, H.; Muldowney, M.T.; Yanagisawa, L.L.; Kutsch, O.; Clements, J.E.; Benveniste, E.N. Suppressor of cytokine signaling 3 inhibits antiviral IFN-β signaling to enhance HIV-1 replication in macrophages. J. Immunol. 2010, 185, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.; Zink, W.; Xiong, H.; Gendelman, H.E. HIV-1-associated dementia: A metabolic encephalopathy perpetrated by virus-infected and immune-competent mononuclear phagocytes. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. 2), S43–S54. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Duan, F.; Morsey, B.; Persidsky, Y.; Kanmogne, G.D. HIV-1 activates proinflammatory and interferon-inducible genes in human brain microvascular endothelial cells: Putative mechanisms of blood-brain barrier dysfunction. J. Int. Soc. Cereb. Blood Flow Metab. 2008, 28, 697–711. [Google Scholar] [CrossRef] [PubMed]
- King, C.A. Kaposi’s sarcoma-associated herpesvirus kaposin B induces unique monophosphorylation of STAT3 at serine 727 and MK2-mediated inactivation of the STAT3 transcriptional repressor TRIM28. J. Virol. 2013, 87, 8779–8791. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2017, 128, 366–375. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, Y.; Wu, C.; Zhang, Y.-J. Interferon Independent Non-Canonical STAT Activation and Virus Induced Inflammation. Viruses 2018, 10, 196. https://doi.org/10.3390/v10040196
Nan Y, Wu C, Zhang Y-J. Interferon Independent Non-Canonical STAT Activation and Virus Induced Inflammation. Viruses. 2018; 10(4):196. https://doi.org/10.3390/v10040196
Chicago/Turabian StyleNan, Yuchen, Chunyan Wu, and Yan-Jin Zhang. 2018. "Interferon Independent Non-Canonical STAT Activation and Virus Induced Inflammation" Viruses 10, no. 4: 196. https://doi.org/10.3390/v10040196
APA StyleNan, Y., Wu, C., & Zhang, Y. -J. (2018). Interferon Independent Non-Canonical STAT Activation and Virus Induced Inflammation. Viruses, 10(4), 196. https://doi.org/10.3390/v10040196