Role of the ERK1/2 Signaling Pathway in the Replication of Junín and Tacaribe Viruses

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Compounds
2.2. Antibodies
2.3. Plasmids
2.4. Viral Adsorption Assay
2.5. Viral Internalization Assay
2.6. Viral Uncoating Assay
2.7. Cell Viability Assay
2.8. Western Blot Analysis
2.9. Immunofluorescence Assay
2.10. Syncytium Formation Assay
2.11. Quantitative RT-PCR
2.12. Replicon Assay
2.13. Cell-Based Translation Assay
2.14. Statistical Analysis
3. Results
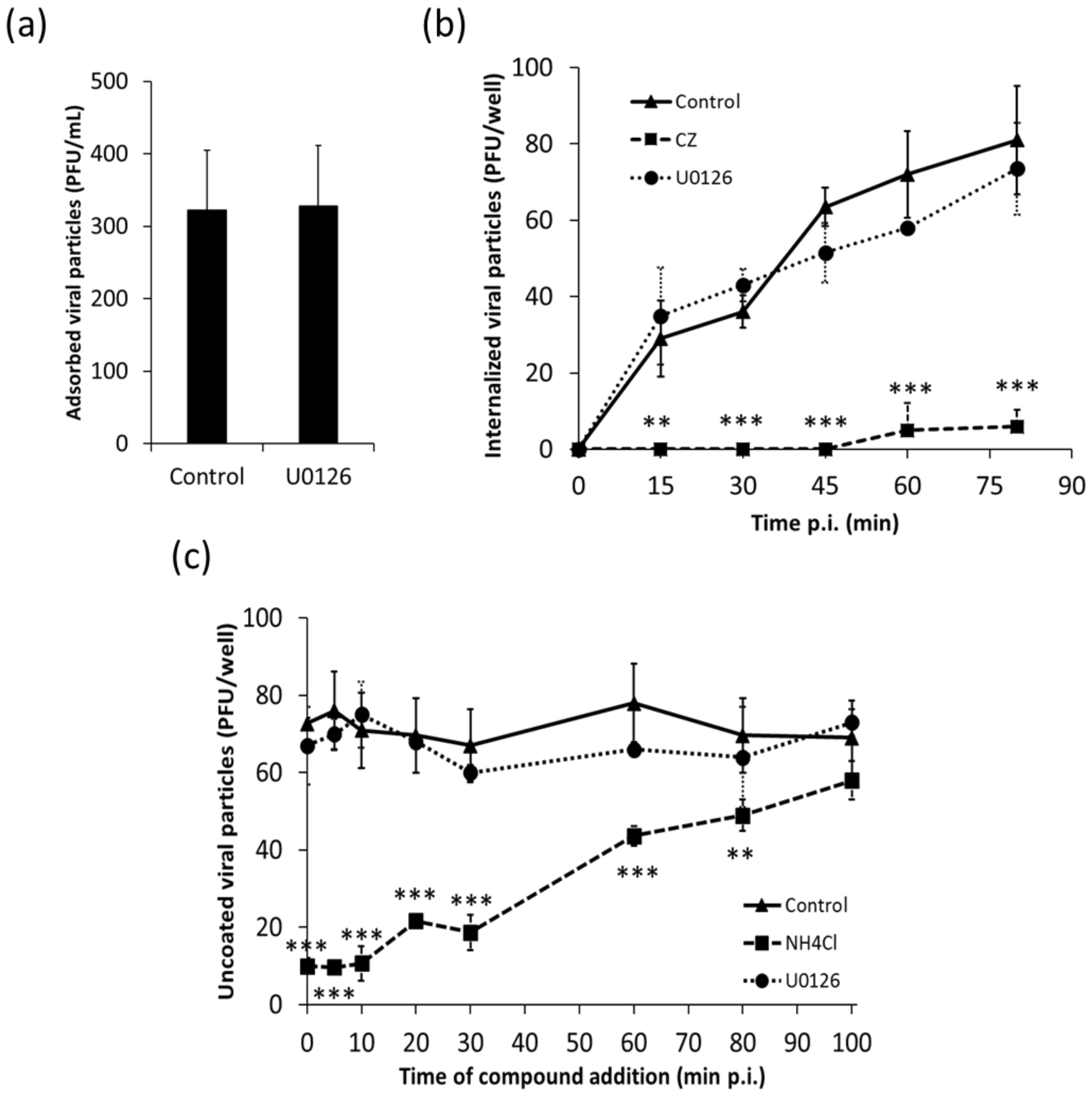
3.1. Effect of U0126 on Initial Steps of Viral Replicative Cycle
3.2. Effect of U0126 on the Expression of JUNV Proteins
3.3. Effect of U0126 on Viral RNA Synthesis
3.4. Effect of U0126 on Viral mRNA Translation
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martinez-Sobrido, L.; de la Torre, J.C. Novel strategies for development of hemorrhagic fever arenavirus live-attenuated vaccines. Expert Rev. Vaccines 2016, 15, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Pasquato, A.; Kunz, S. Novel drug discovery approaches for treating arenavirus infections. Expert Opin. Drug Discov. 2016, 11, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Enria, D.A.; Briggiler, A.M.; Sánchez, Z. Treatment of Argentine hemorrhagic fever. Antivir. Res. 2008, 78, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Linero, F.N.; Sepúlveda, C.S.; Giovannoni, F.; Castilla, V.; García, C.C.; Scolaro, L.A.; Damonte, E.B. Host cell factors as antiviral targets in arenavirus infection. Viruses 2012, 4, 1569–1591. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, D.; Feldmann, H. Vaccines for viral hemorrhagic fevers-progress and shortcomings. Curr. Opin. Virol. 2013, 3, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liao, Y.; Liu, D.X. Regulation of Stress Responses and Translational Control by Coronavirus. Viruses 2016, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Furler, R.L.; Uittenbogaart, C.H. Signaling through the P38 and ERK pathways: A common link between HIV replication and the immune response. Immunol. Res. 2010, 48, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Pleschka, S. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 2008, 389, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.A.; Bruett, L.; Douglass, B.R.; Herbst, D.S.; Zink, M.C.; Clements, J.E. Visna virus-induced activation of MAPK is required for virus replication and correlates with virus-induced neuropathology. J. Virol. 2002, 76, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway—A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Feng, M.; Liao, M.; Cao, W. Inhibition of ERK/MAPK suppresses avian leukosis virus subgroup A and B replication. Microb. Pathog. 2017, 102, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Droebner, K.; Pleschka, S.; Ludwig, S.; Planz, O. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antivir. Res. 2011, 92, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Huynh, V.T.; Lim, Y.S.; Tran, S.C.; Pham, T.M.; Nguyen, L.N.; Hwang, S.B. Hepatitis C Virus Nonstructural 5A Protein Interacts with Abelson Interactor 1 and Modulates Epidermal Growth Factor-mediated MEK/ERK Signaling Pathway. J. Biol. Chem. 2016, 291, 22607–22617. [Google Scholar] [CrossRef] [PubMed]
- Kew, V.; Wills, M.; Reeves, M. HCMV activation of ERK-MAPK drives a multi-factorial response promoting the survival of infected myeloid progenitors. J. Mol. Biochem. 2017, 6, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Wolff, T.; Ehrhardt, C.; Wurzer, W.J.; Reinhardt, J.; Planz, O.; Pleschka, S. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett. 2004, 561, 37–43. [Google Scholar] [CrossRef]
- Luo, H.; Yanagawa, B.; Zhang, J.; Luo, Z.; Zhang, M.; Esfandiarei, M.; Carthy, C.; Wilson, J.E.; Yang, D.; McManus, B.M. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J. Virol. 2002, 76, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Sreekanth, G.P.; Chuncharunee, A.; Sirimontaporn, A.; Panaampon, J.; Srisawat, C.; Morchang, A.; Malakar, S.; Thuwajit, P.; Kooptiwut, S.; Suttitheptumrong, A.; et al. Role of ERK1/2 signaling in dengue virus-induced liver injury. Virus Res. 2014, 188, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.C.; Lai, C.C.; Shiu, S.L.; Chuang, P.H.; Tzou, B.C.; Lin, Y.Y.; Tsai, F.J.; Lin, C.W. Japanese encephalitis virus down-regulates thioredoxin and induces ROS-mediated ASK1-ERK/p38 MAPK activation in human promonocyte cells. Microbes Infect. 2010, 12, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Feng, H.; Luo, L.; Zhou, Q.; Luo, Z.; Peng, Y. Distinct effects of knocking down MEK1 and MEK2 on replication of herpes simplex virus type 2. Virus Res. 2010, 150, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Zhu, M.; Xiong, Q.; Wang, Y.; Xu, C.; Sun, J.; Wang, C.; Zhang, H.; Xu, P.; Peng, Y. Regulation of enterovirus 2A protease-associated viral IRES activities by the cell’s ERK signaling cascade: Implicating ERK as an efficiently antiviral target. Antivir. Res. 2017, 143, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Haasbach, E.; Müller, C.; Ehrhardt, C.; Schreiber, A.; Pleschka, S.; Ludwig, S.; Planz, O. The MEK-inhibitor CI-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antivir. Res. 2017, 142, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.E.; Brunetti, J.E.; Wachsman, M.B.; Scolaro, L.A.; Castilla, V. Raf/MEK/ERK pathway activation is required for Junín virus replication. J. Gen. Virol. 2014, 95, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Pifat, D.Y.; Kenyon, R.H.; Peters, C.J.; McCormick, J.B.; Kiley, M.P. Junin virus monoclonal antibodies: Characterization and cross-reactivity with other arenaviruses. J. Gen. Virol. 1989, 70, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Casabona, J.C.; Levingston Macleod, J.M.; Loureiro, M.E.; Gomez, G.A.; Lopez, N. The RING domain and the L79 residue of Z protein are involved in both the rescue of nucleocapsids and the incorporation of glycoproteins into infectious chimeric arenavirus-like particles. J. Virol. 2009, 83, 7029–7039. [Google Scholar] [CrossRef] [PubMed]
- D’Antuono, A.; Loureiro, M.E.; Foscaldi, S.; Marino-Buslje, C.; Lopez, N. Differential contributions of Tacaribe arenavirus nucleoprotein N-terminal and C-terminal residues to nucleocapsid functional activity. J. Virol. 2014, 88, 6492–6505. [Google Scholar] [CrossRef] [PubMed]
- Franze-Fernández, M.T.; Zetina, C.; Iapalucci, S.; Lucero, M.A.; Bouissou, C.; López, R.; Rey, O.; Daheli, M.; Cohen, G.N.; Zakin, M.M. Molecular structure and early events in the replication of Tacaribe arenavirus S RNA. Virus Res. 1987, 7, 309–324. [Google Scholar] [CrossRef]
- López, N.; Jácamo, R.; Franze-Fernández, M.T. Transcription and RNA replication of tacaribe virus genome and antigenome analogs require N and L proteins: Z protein is an inhibitor of these processes. J. Virol. 2001, 75, 12241–12251. [Google Scholar] [CrossRef] [PubMed]
- Foscaldi, S.; D’Antuono, A.; Noval, M.G.; de Prat Gay, G.; Scolaro, L.; Lopez, N. Regulation of Tacaribe Mammarenavirus Translation: Positive 5′ and Negative 3′ Elements and Role of Key Cellular Factors. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Radecke, F.; Spielhofer, P.; Schneider, H.; Kaelin, K.; Huber, M.; Dötsch, C.; Christiansen, G.; Billeter, M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995, 14, 5773–5784. [Google Scholar] [PubMed]
- Castilla, V.; Larzábal, M.; Sgalippa, N.A.; Wachsman, M.B.; Coto, C.E. Antiviral mode of action of a synthetic brassinosteroid against Junin virus replication. Antivir. Res. 2005, 68, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Corbett, K.D.; Farzan, M.; Choe, H.; Harrison, S.C. Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses. Nat. Struct. Mol. Biol. 2010, 17, 438–444. [Google Scholar] [CrossRef] [PubMed]
- York, J.; Nunberg, J.H. Myristoylation of the Arenavirus Envelope Glycoprotein Stable Signal Peptide Is Critical for Membrane Fusion but Dispensable for Virion Morphogenesis. J. Virol. 2016, 90, 8341–8350. [Google Scholar] [CrossRef] [PubMed]
- Castilla, V.; Mersich, S.E. Low-pH-induced fusion of Vero cells infected with Junin virus. Arch. Virol. 1996, 141, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Powers, L.S.; Gross, T.J.; Flaherty, D.M.; Barrett, C.W.; Hunninghake, G.W. Active ERK contributes to protein translation by preventing JNK-dependent inhibition of protein phosphatase 1. J. Immunol. 2016, 177, 1636–1645. [Google Scholar] [CrossRef]
- Linero, F.N.; Thomas, M.G.; Boccaccio, G.L.; Scolaro, L.A. Junin virus infection impairs stress-granule formation in Vero cells treated with arsenite via inhibition of eIF2α phosphorylation. J. Gen. Virol. 2011, 92, 2889–2899. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.; Hitti, E.; Khabar, K.S.A. ARED-Plus: An updated and expanded database of AU-rich element-containing mRNAs and pre-mRNAs. Nucleic Acids Res. 2018, 46, D218–D220. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liu, Y.; Zhang, X. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J. Virol. 2007, 81, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Alam, M.I.; Ehrhardt, C.; Wagner, R.; Planz, O.; Klenk, H.D.; Ludwig, S.; Pleschka, S. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Calpha-mediated activation of ERK signaling. J. Biol. Chem. 2006, 281, 16707–16715. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell. Microbiol. 2011, 13, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Knebel, A.; Botting, C.H.; Galloway, C.S.; Precious, B.L.; Jackson, D.; Elliott, R.M.; Randall, R.E. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology 2009, 383, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Levingston Macleod, J.M.; D’Antuono, A.; Loureiro, M.E.; Casabona, J.C.; Gomez, G.A.; Lopez, N. Identification of two functional domains within the arenavirus nucleoprotein. J. Virol. 2011, 85, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Knopp, K.A.; Ngo, T.; Gershon, P.D.; Buchmeier, M.J. Single nucleoprotein residue modulates arenavirus replication complex formation. mBio 2015, 6, e00524-15. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Minder, P.; Caì, Y.; Kuhn, J.H.; Yates, J.R., 3rd; Torbett, B.E.; de la Torre, J.C. Interactome analysis of the lymphocytic choriomeningitis virus nucleoprotein in infected cells reveals ATPase Na+/K+ transporting subunit Alpha 1 and prohibitin as host-cell factors involved in the life cycle of mammarenaviruses. PLoS Pathog. 2018, 14, e1006892. [Google Scholar] [CrossRef] [PubMed]
- King, B.R.; Hershkowitz, D.; Eisenhauer, P.L.; Weir, M.E.; Ziegler, C.M.; Russo, J.; Brucea, E.A.; Ballifc, B.A.; Botten, J. A Map of the Arenavirus Nucleoprotein-Host Protein Interactome Reveals that Junín Virus Selectively Impairs the Antiviral Activity of Double-Stranded RNA-Activated Protein Kinase (PKR). J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol. Lett. 2017, 13, 1041–1047. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunetti, J.E.; Foscaldi, S.; Quintana, V.M.; Scolaro, L.A.; López, N.; Castilla, V. Role of the ERK1/2 Signaling Pathway in the Replication of Junín and Tacaribe Viruses. Viruses 2018, 10, 199. https://doi.org/10.3390/v10040199
Brunetti JE, Foscaldi S, Quintana VM, Scolaro LA, López N, Castilla V. Role of the ERK1/2 Signaling Pathway in the Replication of Junín and Tacaribe Viruses. Viruses. 2018; 10(4):199. https://doi.org/10.3390/v10040199
Chicago/Turabian StyleBrunetti, Jesús E., Sabrina Foscaldi, Verónica M. Quintana, Luis A. Scolaro, Nora López, and Viviana Castilla. 2018. "Role of the ERK1/2 Signaling Pathway in the Replication of Junín and Tacaribe Viruses" Viruses 10, no. 4: 199. https://doi.org/10.3390/v10040199
APA StyleBrunetti, J. E., Foscaldi, S., Quintana, V. M., Scolaro, L. A., López, N., & Castilla, V. (2018). Role of the ERK1/2 Signaling Pathway in the Replication of Junín and Tacaribe Viruses. Viruses, 10(4), 199. https://doi.org/10.3390/v10040199