Probing Molecular Insights into Zika Virus–Host Interactions

 , ,
, ,  and
and 
Abstract
:1. Introduction
1.1. The Zika Virus (ZIKV): An Emerging Public Health Threat
1.2. The Organization of Zika Virus
1.3. The Infectious Cycle of ZIKV and Human Transmission
1.4. A Brief History of ZIKV
1.5. What Has Been Learned from the Recent ZIKV Break?
2. Cellular Targets and Viral Entry
2.1. Cellular Targets
2.2. The Cellular Receptors for ZIKV Entry
3. Cellular and Immune Responses to ZIKV Infection
4. Viral Counteraction to Host Antiviral Responses and ZIKV-Induced Cytopathic Effects
4.1. Viral Counteraction to Host Antiviral Responses
4.2. ZIKV-Induced Cytopathic Effects
4.3. The Structural Proteins
4.4. The Non-Structural Proteins
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| ADE | Antibody-dependent enhancement |
| CPE | Cytotoxic effect |
| CNS | Central nerve system |
| DC | Dendritic cell |
| DENV | Dengue virus |
| dsRNA | Double stranded RNA |
| ER | Endoplasmic reticulum |
| HAVcr-1 | Hepatitis A virus cellular receptor 1 |
| hEC | Human epithelial cell |
| hNPC | Human neural progenitor cell |
| hBMEC | Human brain microvascular endothelial cell |
| GBS | Guillain–Barré syndrome |
| IFN | Interferon |
| iPSC | Induced pluripotent stem cell |
| IRF3 | Interferon regulatory factor 3 |
| ISGS | Interferon stimulated genes |
| JEV | Japanese Encephalitis Virus |
| NPCs | Neural progenitor stem cells |
| PAMP | Pathogen-associated molecular pattern |
| PR | Protease |
| PRRs | Pattern recognition receptors |
| RdRP | RNA-dependent RNA polymerase |
| RLRs | RIG-I like receptors |
| sfRNA | Subgenomic flavivirus RNA |
| TGN | Trans-Golgi network |
| TLR3 | Toll-like receptor 3 |
| TOR | Target of rapamycin |
| WHO | World health organization |
| WNV | West Nile Virus |
| ZIKV | Zika virus |
References
- Zhao, Z.; Yang, M.; Azar, S.R.; Soong, L.; Weaver, S.C.; Sun, J.; Chen, Y.; Rossi, S.L.; Cai, J. Viral retinopathy in experimental models of zika infection. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4355–4365. [Google Scholar] [CrossRef] [PubMed]
- Sahiner, F.; Sig, A.K.; Savasci, U.; Tekin, K.; Akay, F. Zika virus-associated ocular and neurologic disorders: The emergence of new evidence. Pediatr. Infect. Dis. J. 2017, 36, e341–e346. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.W.; Mackenzie, J. Zika virus and Guillain-Barre syndrome: Another viral cause to add to the list. Lancet 2016, 387, 1486–1488. [Google Scholar] [CrossRef]
- Wen, Z.; Song, H.; Ming, G.L. How does Zika virus cause microcephaly? Genes Dev. 2017, 31, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Carod-Artal, F.J. Neurological complications of Zika virus infection. Expert Rev. Anti-Infect. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- De Paula Freitas, B.; de Oliveira Dias, J.R.; Prazeres, J.; Sacramento, G.A.; Ko, A.I.; Maia, M.; Belfort, R., Jr. Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in salvador, Brazil. JAMA Ophthalmol. 2016, 134, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Faye, O.; Freire, C.C.; Iamarino, A.; Faye, O.; de Oliveira, J.V.; Diallo, M.; Zanotto, P.M.; Sall, A.A. Molecular evolution of Zika virus during its emergence in the 20th century. PLoS Negl. Trop. Dis. 2014, 8, e2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Poulsen, M.; Fenyvuesvolgyi, C.; Yashiroda, Y.; Yoshida, M.; Simard, J.M.; Gallo, R.C.; Zhao, R.Y. Characterization of cytopathic factors through genome-wide analysis of the zika viral proteins in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E376–E385. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.; Holden, K.L.; Edgil, D.; Polacek, C.; Clyde, K. Molecular biology of flaviviruses. Novartis Found. Symp. 2006, 277, 23–39, discussion 40, 71–23, 251–253. [Google Scholar] [PubMed]
- Lobigs, M.; Lee, E.; Ng, M.L.; Pavy, M.; Lobigs, P. A flavivirus signal peptide balances the catalytic activity of two proteases and thereby facilitates virus morphogenesis. Virology 2010, 401, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- Amberg, S.M.; Nestorowicz, A.; McCourt, D.W.; Rice, C.M. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: In vitro and in vivo studies. J. Virol. 1994, 68, 3794–3802. [Google Scholar] [PubMed]
- Lin, J.C.; Lin, S.C.; Chen, W.Y.; Yen, Y.T.; Lai, C.W.; Tao, M.H.; Lin, Y.L.; Miaw, S.C.; Wu-Hsieh, B.A. Dengue viral protease interaction with NF-κB inhibitor α/β results in endothelial cell apoptosis and hemorrhage development. J. Immunol. 2014, 193, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar] [PubMed]
- Elshuber, S.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 2003, 84, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, H.S.; Pauza, C.D.; Bukrinsky, M.; Zhao, R.Y. Roles of HIV-1 auxiliary proteins in viral pathogenesis and host-pathogen interactions. Cell Res. 2005, 15, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Melino, S.; Fucito, S.; Campagna, A.; Wrubl, F.; Gamarnik, A.; Cicero, D.O.; Paci, M. The active essential CFNS3d protein complex. FEBS J. 2006, 273, 3650–3662. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barre syndrome outbreak associated with Zika virus infection in French polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Goertz, G.P.; Abbo, S.R.; Fros, J.J.; Pijlman, G.P. Functional RNA during Zika virus infection. Virus Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chan, J.F.; Tee, K.M.; Choi, G.K.; Lau, S.K.; Woo, P.C.; Tse, H.; Yuen, K.Y. Comparative genomic analysis of pre-epidemic and epidemic zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg. Microbes Infect. 2016, 5, e22. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.B. Zika virus outside Africa. Emerg. Infect. Dis. 2009, 15, 1347–1350. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Higgs, S.; Horne, K.M.; Vanlandingham, D.L. Flavivirus-mosquito interactions. Viruses 2014, 6, 4703–4730. [Google Scholar] [CrossRef] [PubMed]
- Foy, B.D.; Kobylinski, K.C.; Chilson Foy, J.L.; Blitvich, B.J.; Travassos da Rosa, A.; Haddow, A.D.; Lanciotti, R.S.; Tesh, R.B. Probable non-vector-borne transmission of zika virus, Colorado, USA. Emerg. Infect. Dis. 2011, 17, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Mead, P.S.; Hills, S.L.; Brooks, J.T. Zika virus as a sexually transmitted pathogen. Curr. Opin. Infect. Dis. 2018, 31, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Nhan, T.; Robin, E.; Roche, C.; Bierlaire, D.; Zisou, K.; Shan Yan, A.; Cao-Lormeau, V.M.; Broult, J. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Eurosurveillance 2014, 19. [Google Scholar] [CrossRef]
- Govero, J.; Esakky, P.; Scheaffer, S.M.; Fernandez, E.; Drury, A.; Platt, D.J.; Gorman, M.J.; Richner, J.M.; Caine, E.A.; Salazar, V.; et al. Zika virus infection damages the testes in mice. Nature 2016, 540, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Li, S.; Ma, S.; Jia, L.; Zhang, F.; Zhang, Y.; Zhang, J.; Wong, G.; Zhang, S.; Lu, X.; et al. Zika virus causes testis damage and leads to male infertility in mice. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed]
- Joguet, G.; Mansuy, J.M.; Matusali, G.; Hamdi, S.; Walschaerts, M.; Pavili, L.; Guyomard, S.; Prisant, N.; Lamarre, P.; Dejucq-Rainsford, N.; et al. Effect of acute Zika virus infection on sperm and virus clearance in body fluids: A prospective observational study. Lancet Infect. Dis. 2017, 17, 1200–1208. [Google Scholar] [CrossRef]
- Cao, B.; Diamond, M.S.; Mysorekar, I.U. Maternal-fetal transmission of zika virus: Routes and signals for infection. J. Interferon Cytokine Res. 2017, 37, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; de Noronha, L.; Duarte Dos Santos, C.N. Maternal-fetal transmission of the zika virus: An intriguing interplay. Tissue Barriers 2018, 6, e1402143. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika virus associated with microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Dick, G.W. Zika virus. II. Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef]
- Bell, T.M.; Field, E.J.; Narang, H.K. Zika virus infection of the central nervous system of mice. Arch. Gesamte Virusforsch. 1971, 35, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, federated states of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Ioos, S.; Mallet, H.P.; Leparc Goffart, I.; Gauthier, V.; Cardoso, T.; Herida, M. Current Zika virus epidemiology and recent epidemics. Med. Mal. Infect. 2014, 44, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Victora, C.G.; Schuler-Faccini, L.; Matijasevich, A.; Ribeiro, E.; Pessoa, A.; Barros, F.C. Microcephaly in Brazil: How to interpret reported numbers? Lancet 2016, 387, 621–624. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Haddow, A.D.; Schuh, A.J.; Yasuda, C.Y.; Kasper, M.R.; Heang, V.; Huy, R.; Guzman, H.; Tesh, R.B.; Weaver, S.C. Genetic characterization of Zika virus strains: Geographic expansion of the Asian lineage. PLoS Negl. Trop. Dis. 2012, 6, e1477. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; DeMaso, C.R.; Pelc, R.S.; Speer, S.D.; Smith, A.R.Y.; Goo, L.; Platt, D.J.; Mascola, J.R.; Graham, B.S.; Mulligan, M.J.; et al. Broadly neutralizing activity of Zika virus-immune sera identifies a single viral serotype. Cell Rep. 2016, 16, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Anfasa, F.; Siegers, J.Y.; van der Kroeg, M.; Mumtaz, N.; Stalin Raj, V.; de Vrij, F.M.S.; Widagdo, W.; Gabriel, G.; Salinas, S.; Simonin, Y.; et al. Phenotypic differences between Asian and African lineage Zika viruses in human neural progenitor cells. MSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, C.; Liu, S.; Zhou, H.; Yang, K.; Zhao, Q.; Ji, X.; Chen, C.; Xie, W.; Wang, Z.; et al. A mutation identified in neonatal microcephaly destabilizes Zika virus NS1 assembly in vitro. Sci Rep. 2017, 7, 42580. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Du, S.; Shan, C.; Nie, K.; Zhang, R.; Li, X.F.; Zhang, R.; Wang, T.; Qin, C.F.; et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 2017, 545, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Sariol, C.A.; Nogueira, M.L.; Vasilakis, N. A tale of two viruses: Does heterologous flavivirus immunity enhance Zika disease? Trends Microbiol. 2018, 26, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Kawiecki, A.B.; Christofferson, R.C. Zika virus-induced antibody response enhances dengue virus serotype 2 replication in vitro. J. Infect. Dis. 2016, 214, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Valiant, W.G.; Mattapallil, M.J.; Walker, M.; Huang, Y.S.; Vanlandingham, D.L.; Misamore, J.; Greenhouse, J.; Weiss, D.E.; Verthelyi, D.; et al. Prior exposure to Zika virus significantly enhances peak Dengue-2 viremia in Rhesus macaques. Sci. Rep. 2017, 7, 10498. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Supasa, P.; Wongwiwat, W.; Rouvinski, A.; Barba-Spaeth, G.; Duangchinda, T.; Sakuntabhai, A.; Cao-Lormeau, V.M.; Malasit, P.; Rey, F.A.; et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with Zika virus. Nat. Immunol. 2016, 17, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.M.; Carlin, E.R.; Jenkins, M.M.; Tan, A.L.; Barcellona, C.M.; Nicholson, C.O.; Michael, S.F.; Isern, S. Dengue virus antibodies enhance Zika virus infection. Clin. Transl. Immunol. 2016, 5, e117. [Google Scholar] [CrossRef] [PubMed]
- Londono-Renteria, B.; Troupin, A.; Cardenas, J.C.; Hall, A.; Perez, O.G.; Cardenas, L.; Hartstone-Rose, A.; Halstead, S.B.; Colpitts, T.M. A relevant in vitro human model for the study of Zika virus antibody-dependent enhancement. J. Gen. Virol. 2017, 98, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Dengue may bring out the worst in Zika. Science 2017, 355, 1362. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.S.; Miller, A.; Sapparapu, G.; Fernandez, E.; Klose, T.; Long, F.; Fokine, A.; Porta, J.C.; Jiang, W.; Diamond, M.S.; et al. A human antibody against Zika virus crosslinks the e protein to prevent infection. Nat. Commun. 2017, 8, 14722. [Google Scholar] [CrossRef] [PubMed]
- Cugola, F.R.; Fernandes, I.R.; Russo, F.B.; Freitas, B.C.; Dias, J.L.; Guimaraes, K.P.; Benazzato, C.; Almeida, N.; Pignatari, G.C.; Romero, S.; et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Herrlinger, S.; Zhu, Y.N.; Yang, M.; Goodfellow, F.; Stice, S.L.; Qi, X.P.; Brindley, M.A.; Chen, J.F. The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 2017, 144, 4114–4124. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, D.; Ye, Q.; Hong, S.; Jiang, Y.; Liu, X.; Zhang, N.; Shi, L.; Qin, C.F.; Xu, Z. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 2016, 19, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, H.J.; Wang, Q.; Liu, Z.Y.; Yuan, L.; Huang, X.Y.; Li, G.; Ye, Q.; Yang, H.; Shi, L.; et al. American strain of Zika virus causes more severe microcephaly than an old Asian strain in neonatal mice. EBioMedicine 2017, 25, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Herrlinger, S.; Yang, S.L.; Lai, F.; Moore, J.M.; Brindley, M.A.; Chen, J.F. Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 2016, 143, 4127–4136. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika virus infection in human skin cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.R.; Zimmerman, M.G.; Suthar, M.S. Taking the defensive: Immune control of Zika virus infection. Virus Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hua, S.; Chen, H.R.; Ouyang, Z.; Einkauf, K.; Tse, S.; Ard, K.; Ciaranello, A.; Yawetz, S.; Sax, P.; et al. Transcriptional changes during naturally acquired Zika virus infection render dendritic cells highly conducive to viral replication. Cell Rep. 2017, 21, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Bowen, J.R.; Johnson, E.L.; McDonald, C.E.; Ma, H.; O’Neal, J.T.; Rajakumar, A.; Wrammert, J.; Rimawi, B.H.; Pulendran, B.; et al. Zika virus infects human placental macrophages. Cell Host Microbe 2016, 20, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Mladinich, M.C.; Schwedes, J.; Mackow, E.R. Zika virus persistently infects and is basolaterally released from primary human brain microvascular endothelial cells. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Yip, C.C.; Tsang, J.O.; Tee, K.M.; Cai, J.P.; Chik, K.K.; Zhu, Z.; Chan, C.C.; Choi, G.K.; Sridhar, S.; et al. Differential cell line susceptibility to the emerging Zika virus: Implications for disease pathogenesis, non-vector-borne human transmission and animal reservoirs. Emerg. Microbes Infect. 2016, 5, e93. [Google Scholar] [CrossRef] [PubMed]
- Thepparit, C.; Smith, D.R. Serotype-specific entry of dengue virus into liver cells: Identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J. Virol. 2004, 78, 12647–12656. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Burstyn-Cohen, T. Tam receptors and the clearance of apoptotic cells. Ann. N. Y. Acad. Sci. 2010, 1209, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vallejo, J.J.; Ambrosini, M.; Overbeek, A.; van Riel, W.E.; Bloem, K.; Unger, W.W.; Chiodo, F.; Bolscher, J.G.; Nazmi, K.; Kalay, H.; et al. Multivalent glycopeptide dendrimers for the targeted delivery of antigens to dendritic cells. Mol. Immunol. 2013, 53, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Stroher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J. Virol. 2012, 86, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for zaire ebolavirus and lake victoria marburgvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 8426–8431. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Chen, I.S. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 2014, 88, 4275–4290. [Google Scholar] [CrossRef] [PubMed]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. AXL mediates Zika virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Mancia Leon, W.R.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Wang, C.; Fang-Hoover, J.; Harris, E.; Pereira, L. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe 2016, 20, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.A.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika virus infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Wang, Z.; Zhen, Z.D.; Feng, K.H.; Guo, J.; Gao, N.; Fan, D.Y.; Han, D.S.; Wang, P.G.; An, J. AXL is not an indispensable factor for Zika virus infection in mice. J. Gen. Virol. 2017, 98, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Ferraris, P.; Wichit, S.; Diop, F.; Talignani, L.; Pompon, J.; Garcia, D.; Liegeois, F.; Sall, A.A.; Yssel, H.; et al. African and Asian Zika virus strains differentially induce early antiviral responses in primary human astrocytes. Infect. Genet. Evol. 2017, 49, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Stefanik, M.; Formanova, P.; Bily, T.; Vancova, M.; Eyer, L.; Palus, M.; Salat, J.; Braconi, C.T.; Zanotto, P.M.A.; Gould, E.A.; et al. Characterisation of Zika virus infection in primary human astrocytes. BMC Neurosci 2018, 19, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, Y.F.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.L.; Cui, Y.R.; Bai, R.; Liang, Y.J.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type i interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Persaud, M.; Martinez-Lopez, A.; Buffone, C.; Porcelli, S.A.; Diaz-Griffero, F. Infection by zika viruses requires the transmembrane protein AXL, endocytosis and low ph. Virology 2018, 518, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.S.; Chen, W.; Chan, Y.; Bowman, J.W.; Chang, L.C.; Choi, Y.; Yoo, J.S.; Ge, J.; Cheng, G.; Bonnin, A.; et al. Asian Zika virus strains target CD14+ blood monocytes and induce M2-skewed immunosuppression during pregnancy. Nat. Microbiol. 2017, 2, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Michlmayr, D.; Andrade, P.; Gonzalez, K.; Balmaseda, A.; Harris, E. CD14+ CD16+ monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat. Microbiol. 2017, 2, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Lum, F.M.; Lee, D.; Chua, T.K.; Tan, J.J.L.; Lee, C.Y.P.; Liu, X.; Fang, Y.; Lee, B.; Yee, W.X.; Rickett, N.Y.; et al. Zika virus infection preferentially counterbalances human peripheral monocyte and/or NK cell activity. MSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.Y.; Gao, N.; Wang, Z.Y.; Cui, X.Y.; Zhou, D.S.; Fan, D.Y.; Chen, H.; Wang, P.G.; An, J. Sertoli cells are susceptible to zikv infection in mouse testis. Front. Cell. Infect. Microbiol. 2017, 7, 272. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.N.; Strange, D.P.; Maharaj, P.N.; Shi, P.Y.; Verma, S. Zika virus infects human sertoli cells and modulates the integrity of the in vitro blood-testis barrier model. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Salam, A.P.; Horby, P. Isolation of viable Zika virus from spermatozoa. Lancet Infect. Dis. 2018, 18, 144. [Google Scholar] [CrossRef]
- Bagasra, O.; Addanki, K.C.; Goodwin, G.R.; Hughes, B.W.; Pandey, P.; McLean, E. Cellular targets and receptor of sexual transmission of zika virus. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Alcendor, D.J. Zika virus infection of the human glomerular cells: Implications for viral reservoirs and renal pathogenesis. J. Infect. Dis. 2017, 216, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Roach, T.; Alcendor, D.J. Zika virus infection of cellular components of the blood-retinal barriers: Implications for viral associated congenital ocular disease. J. Neuroinflamm. 2017, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. Cytoarchitecture of Zika virus infection in human neuroblastoma and aedes albopictus cell lines. Virology 2017, 501, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Bos, S.; Viranaicken, W.; Turpin, J.; El-Kalamouni, C.; Roche, M.; Krejbich-Trotot, P.; Despres, P.; Gadea, G. The structural proteins of epidemic and historical strains of Zika virus differ in their ability to initiate viral infection in human host cells. Virology 2018, 516, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Frumence, E.; Roche, M.; Krejbich-Trotot, P.; El-Kalamouni, C.; Nativel, B.; Rondeau, P.; Misse, D.; Gadea, G.; Viranaicken, W.; Despres, P. The south pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN-β production and apoptosis induction. Virology 2016, 493, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Colavita, F.; Musumeci, G.; Caglioti, C. Human osteoblast-like cells are permissive for Zika virus replication. J. Rheumatol. 2018, 45, 443. [Google Scholar] [CrossRef] [PubMed]
- Tappe, D.; Perez-Giron, J.V.; Zammarchi, L.; Rissland, J.; Ferreira, D.F.; Jaenisch, T.; Gomez-Medina, S.; Gunther, S.; Bartoloni, A.; Munoz-Fontela, C.; et al. Cytokine kinetics of zika virus-infected patients from acute to reconvalescent phase. Med. Microbiol. Immunol. 2016, 205, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Pham, J.; Sidney, J.; O’Rourke, P.H.; Paul, S.; Peters, B.; Martini, S.R.; de Silva, A.D.; Ricciardi, M.J.; Magnani, D.M.; et al. Prior dengue virus exposure shapes t cell immunity to Zika virus in humans. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Elong Ngono, A.; Shresta, S. Immune response to dengue and zika. Annu. Rev. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.A.; Stiasny, K.; Vaney, M.C.; Dellarole, M.; Heinz, F.X. The bright and the dark side of human antibody responses to flaviviruses: Lessons for vaccine design. EMBO Rep. 2018, 19, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Jeng, S.; McWeeney, S.K.; Hirsch, A.J. A microrna screen identifies the wnt signaling pathway as a regulator of the interferon response during flavivirus infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, C.; Lindqvist, R.; Kurhade, C.; Vonderstein, K.; Pasto, J.; Edlund, K.; Upadhyay, A.S.; Overby, A.K. Viperin restricts Zika virus and tick-borne encephalitis virus replication by targeting NS3 for proteasomal degradation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Anglero-Rodriguez, Y.I.; MacLeod, H.J.; Kang, S.; Carlson, J.S.; Jupatanakul, N.; Dimopoulos, G. Aedes aegypti molecular responses to Zika virus: Modulation of infection by the toll and Jak/Stat immune pathways and virus host factors. Front. Microbiol. 2017, 8, 2050. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.; Delmiro, A.; Blazquez, A.; Ugalde, C.; Arenas, J.; Martin, M.A. Bulk autophagy, but not mitophagy, is increased in cellular model of mitochondrial disease. Biochim. Biophys. Acta 2014, 1842, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Tetro, J.A. Zika and microcephaly: Causation, correlation, or coincidence? Microbes Infect. 2016, 18, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lennemann, N.J.; Coyne, C.B. Dengue and zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Weisman, R.; Roitburg, I.; Schonbrun, M.; Harari, R.; Kupiec, M. Opposite effects of TOR1 and TOR2 on nitrogen starvation responses in fission yeast. Genetics 2007, 175, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Fenyvuesvolgyi, C.; Elder, R.T.; Benko, Z.; Liang, D.; Zhao, R.Y. Fission yeast homologue of Tip41-like proteins regulates type 2A phosphatases and responses to nitrogen sources. Biochim. Biophys. Acta 2005, 1746, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Elong Ngono, A.; Vizcarra, E.A.; Tang, W.W.; Sheets, N.; Joo, Y.; Kim, K.; Gorman, M.J.; Diamond, M.S.; Shresta, S. Mapping and role of the CD8+ T cell response during primary Zika virus infection in mice. Cell Host Microbe 2017, 21, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Zammarchi, L.; Tappe, D.; Fortuna, C.; Remoli, M.E.; Gunther, S.; Venturi, G.; Bartoloni, A.; Schmidt-Chanasit, J. Zika virus infection in a traveller returning to Europe from Brazil, March 2015. Eurosurveillance 2015, 20, 21153. [Google Scholar] [CrossRef] [PubMed]
- Andrade, D.V.; Harris, E. Recent advances in understanding the adaptive immune response to Zika virus and the effect of previous flavivirus exposure. Virus Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bardina, S.V.; Bunduc, P.; Tripathi, S.; Duehr, J.; Frere, J.J.; Brown, J.A.; Nachbagauer, R.; Foster, G.A.; Krysztof, D.; Tortorella, D.; et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 2017, 356, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Priyamvada, L.; Quicke, K.M.; Hudson, W.H.; Onlamoon, N.; Sewatanon, J.; Edupuganti, S.; Pattanapanyasat, K.; Chokephaibulkit, K.; Mulligan, M.J.; Wilson, P.C.; et al. Human antibody responses after dengue virus infection are highly cross-reactive to zika virus. Proc. Natl. Acad. Sci. USA 2016, 113, 7852–7857. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A mouse model of zika virus pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Tesh, R.B.; Azar, S.R.; Muruato, A.E.; Hanley, K.A.; Auguste, A.J.; Langsjoen, R.M.; Paessler, S.; Vasilakis, N.; Weaver, S.C. Characterization of a novel murine model to study zika virus. Am. J. Trop. Med. Hyg. 2016, 94, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika virus targets human Stat2 to inhibit type I interferon signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.; Yuen, K.S.; Chan, J.F.; Chan, C.P.; Wang, P.H.; Cai, J.P.; Zhang, S.; Liang, M.; Kok, K.H.; Chan, C.P.; et al. Selective activation of type ii interferon signaling by Zika virus NS5 protein. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.N.; Qian, X.; Song, H.; Ming, G.L. Neural stem cells attacked by zika virus. Cell Res. 2016, 26, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Slomnicki, L.P.; Chung, D.H.; Parker, A.; Hermann, T.; Boyd, N.L.; Hetman, M. Ribosomal stress and Tp53-mediated neuronal apoptosis in response to capsid protein of the Zika virus. Sci. Rep. 2017, 7, 16652. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.J.; Song, G.; Qian, X.; Pan, J.; Xu, D.; Rho, H.S.; Kim, N.S.; Habela, C.; Zheng, L.; Jacob, F.; et al. Zika-virus-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins. Cell Stem Cell 2017, 21, 349–358 e346. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.Y.; Liu, Z.Y.; Nian, Q.G.; Zhu, L.; Wang, N.; Deng, Y.Q.; Zhao, H.; Ji, X.; Li, X.F.; Wang, X.; et al. A single residue in the alphab helix of the e protein is critical for Zika virus thermostability. Emerg. Microbes Infect. 2018, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Brault, A.C.; Domi, A.; McDonald, E.M.; Talmi-Frank, D.; McCurley, N.; Basu, R.; Robinson, H.L.; Hellerstein, M.; Duggal, N.K.; Bowen, R.A.; et al. A zika vaccine targeting NS1 protein protects immunocompetent adult mice in a lethal challenge model. Sci. Rep. 2017, 7, 14769. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M. Yeast help identify cytopathic factors of zika virus. Cell Biosci. 2017, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K. A prize for proliferation. Cell 2001, 107, 689–701. [Google Scholar] [CrossRef]
- Nurse, P. Cyclin dependent kinases and cell cycle control (nobel lecture). Chembiochem 2002, 3, 596–603. [Google Scholar] [CrossRef]
- Tooze, S.A.; Dikic, I. Autophagy captures the Nobel Prize. Cell 2016, 167, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y. Yeast for virus research. Microb. Cell 2017, 4, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Elder, R.T.; Chen, M.; Cao, J. Fission yeast expression vectors adapted for positive identification of gene insertion and green fluorescent protein fusion. Biotechniques 1998, 25, 438–440, 442, 444. [Google Scholar] [PubMed]
- Li, G.; Zhao, R.Y. Molecular cloning and characterization of small viral genome in fission yeast. Methods Mol. Biol. 2018, 1721, 47–61. [Google Scholar] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic reticulum: The favorite intracellular niche for viral replication and assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Kaufusi, P.H.; Kelley, J.F.; Yanagihara, R.; Nerurkar, V.R. Induction of endoplasmic reticulum-derived replication-competent membrane structures by West Nile virus non-structural protein 4B. PLoS ONE 2014, 9, e84040. [Google Scholar] [CrossRef] [PubMed]
- Sangiambut, S.; Keelapang, P.; Aaskov, J.; Puttikhunt, C.; Kasinrerk, W.; Malasit, P.; Sittisombut, N. Multiple regions in dengue virus capsid protein contribute to nuclear localization during virus infection. J. Gen. Virol. 2008, 89, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Stock, N.K.; Escadafal, C.; Achazi, K.; Cisse, M.; Niedrig, M. Development and characterization of polyclonal peptide antibodies for the detection of yellow fever virus proteins. J. Virol. Methods 2015, 222, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.D.; Stanton, R.A.; Schinazi, R.F. Predicting Zika virus structural biology: Challenges and opportunities for intervention. Antivir. Chem. Chemother. 2015, 24, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Lim, E.X.; Zhang, S.; Fibriansah, G.; Ng, T.S.; Ooi, J.S.; Shi, J.; Lok, S.M. Structure of the thermally stable zika virus. Nature 2016, 533, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Garfias, C.R.; Shan, C.; Luo, H.; Muruato, A.E.; Medeiros, D.B.A.; Mays, E.; Xie, X.; Zou, J.; Roundy, C.M.; Wakamiya, M.; et al. Functional analysis of glycosylation of Zika virus envelope protein. Cell Rep. 2017, 21, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, A.S.; Pattnaik, A.; Sahoo, B.R.; Muthukrishnan, E.; Natarajan, S.K.; Steffen, D.; Vu, H.L.X.; Delhon, G.; Osorio, F.A.; Petro, T.M.; et al. Zika virus encoding non-glycosylated envelope protein is attenuated and defective in neuroinvasion. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Goo, L.; DeMaso, C.R.; Pelc, R.S.; Ledgerwood, J.E.; Graham, B.S.; Kuhn, R.J.; Pierson, T.C. The Zika virus envelope protein glycan loop regulates virion antigenicity. Virology 2018, 515, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Gallichotte, E.N.; Dinnon, K.H., 3rd; Lim, X.N.; Ng, T.S.; Lim, E.X.Y.; Menachery, V.D.; Lok, S.M.; Baric, R.S. CD-loop extension in Zika virus envelope protein key for stability and pathogenesis. J. Infect. Dis. 2017, 216, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Huang, X.Y.; Liu, Z.Y.; Zhang, F.; Zhu, X.L.; Yu, J.Y.; Ji, X.; Xu, Y.P.; Li, G.; Li, C.; et al. A single mutation in the prm protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D.; et al. An infectious cdna clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Mahawaththa, M.C.; Pearce, B.J.G.; Szabo, M.; Graham, B.; Klein, C.D.; Nitsche, C.; Otting, G. Solution conformations of a linked construct of the Zika virus NS2B-NS3 protease. Antiviral Res. 2017, 142, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Phoo, W.W.; Li, Y.; Zhang, Z.; Lee, M.Y.; Loh, Y.R.; Tan, Y.B.; Ng, E.Y.; Lescar, J.; Kang, C.; Luo, D. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 2016, 7, 13410. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ren, J.; Nocadello, S.; Rice, A.J.; Ojeda, I.; Light, S.; Minasov, G.; Vargas, J.; Nagarathnam, D.; Anderson, W.F.; et al. Identification of novel small molecule inhibitors against NS2B/NS3 serine protease from zika virus. Antiviral Res. 2017, 139, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Shafee, N.; AbuBakar, S. Dengue virus type 2 NS3 protease and NS2B-NS3 protease precursor induce apoptosis. J. Gen. Virol. 2003, 84, 2191–2195. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.C.; Shiu, S.L.; Chuang, P.H.; Lin, Y.J.; Wan, L.; Lan, Y.C.; Lin, C.W. Japanese encephalitis virus NS2B-NS3 protease induces caspase 3 activation and mitochondria-mediated apoptosis in human medulloblastoma cells. Virus Res. 2009, 143, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.; Sharma, N.; Singh, S.K. Flavivirus ns1: A multifaceted enigmatic viral protein. Virol. J. 2016, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Viranaicken, W.; Ndebo, A.; Bos, S.; Souque, P.; Gadea, G.; El-Kalamouni, C.; Krejbich-Trotot, P.; Charneau, P.; Despres, P.; Roche, M. Recombinant zika ns1 protein secreted from vero cells is efficient for inducing production of immune serum directed against NS1 dimer. Int. J. Mol. Sci. 2017, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.C.M.; Lamarino, A.; Neto, D.F.L.N.; Sall, A.A.; Zanotto, P.M.A. Spread of the pandemic Zika virus lineage is associated with NS1 codon usage adaptation in humans. BioRxiv 2015. [Google Scholar] [CrossRef]
- Delatorre, E.; Mir, D.; Bello, G. Tracing the origin of the NS1 A188V substitution responsible for recent enhancement of Zika virus Asian genotype infectivity. Mem. Inst. Oswaldo Cruz 2017, 112, 793–795. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ZIKV Strain | Model Used | Host/Location/Year | Microcephaly-Like Phenotypes | Reference |
|---|---|---|---|---|
| Human fetal tissue or organoid models | ||||
| MR766 | Human brain-specific organoids | Rhesus monkey/Uganda/1947 | Increased cell death and reduced proliferation, resulting in decreased neuronal cell-layer volume resembling microcephaly. | [40] |
| MR766 | Human neurospheres and organoids | Rhesus monkey/Uganda/1947 | Growth impairment of neurospheres and organoids | [43] |
| MR766 | Human cerebral organoids | Rhesus monkey/Uganda/1947 | Reduction of organoid growth and volume reminiscent of microcephaly via induction of TLR3 | [57] |
| FSS 13025 | Human brain-specific organoids | Human/Cambodia/2010 | Increased cell death and reduced proliferation, resulting in decreased neuronal cell-layer volume resembling microcephaly. | [40] |
| ZIKV(BR) | Human organoids | Human/Brazil/2015 | Reduction of proliferative zones and disrupted cortical layers; induction of apoptosis, autophagy and impaired neurodevelopment | [59] |
| KU527068 | Aborted human fetal brain | Human/Brazil/2016 | Microcephaly with calcification in the fetal brain and placenta | [32] |
| FB_GWUH | Aborted human fetal brain | Human/USA/2016 | Fetal brain abnormalities with diffuse cerebral cortical thinning | [39] |
| Mouse models | ||||
| PF/2013/KD507 | Mouse | Human/French Polynesia/2013 | Fetal demise or intrauterine growth restriction | [33] |
| ZIKV(BR) | Mouse | Human/Brazil/2015 | Intrauterine growth restriction, including signs of microcephaly and vertical transmission | [59] |
| SZ01 | Mouse vertical transmission | Human/Samoa/2016 | Infection of radial glia cells of dorsal ventricular zone of the fetuses resulting in reduced cavity of lateral ventricles and decreased cortical surface area | [40] |
| SZ01 | Embryonic mouse brain | Human/Samoa/2016 | Cell cycle arrest, apoptosis, and inhibition of NPC differentiation, resulting in cortical thinning and microcephaly | [61] |
| CAM/2010AndVEN/2016 | Neonatal mouse brain | Human/Cambodia/2010 Human/Venezuela/2016 | Neonatal ZIKV infection of VEN/2016 leads to more severe microcephaly than CAM/2010. VEN/2016 strain infection leads to stronger immune response, more severe calcification, more neuronal death and abolished oligodendrocyte development, but less activation of microglial cells. | [62] |
| Primary Cell | Receptor | References | |
|---|---|---|---|
| Brain | |||
| Neural progenitor cells (NPCs) | AXL, TLR3 | [80,84,85] | |
| Astroglial cells | AXL | [36,81,86,87,88] | |
| Microglial cells | AXL | [81] | |
| Placenta | |||
| Hofbauer cells | AXL, Tyro3, TIM1 | [67,68,83] | |
| Trophoblasts | AXL, Tyro3, TIM1, TLR3, TLR8 | [67,68,83] | |
| Endothelial cells | AXL, Tyro3, TIM1 | [33,83] | |
| Skin | |||
| Dermal fibroblasts | AXL, TIM-1, TYRO3, TLR3, RIG-I, MDA5 | [64,89] | |
| Epidermal keratinocytes | AXL, TIM-1, TYRO3, TLR3, RIG-I, MDA5 | [64] | |
| Immune cells | |||
| Immature dendritic cells | DC-SIGN | [64,65] | |
| Dendritic cells | DC-SIGN | [66] | |
| CD14+ monocytes | Unknown | [90,91,92] | |
| CD14+CD16+ monocytes | Unknown | [91] | |
| Testis | |||
| Sertoli cell | AXL | [28,93,94] | |
| Spermatozoa | Tyro3 | [95,96] | |
| Kidney | |||
| Renal mesangial cell | Unknown | [97] | |
| Glomerular podocytes | Unknown | ||
| Renal Glomerular Endothelial Cell | Unknown | ||
| Retina | |||
| Retinal pericytes | Tyro3, AXL | [1,98] | |
| Retinal microvascular endothelial cells | Tyro3, AXL | ||
| Permissive human cell lines | |||
| Cell line | Origins | Permissiveness | References |
| SK-N-SH | Brain/Bone marrow | ** | [99] |
| SH-SY5Y | Nerve | ** | [100] |
| SF268 | CNS in brain | *** | [42,70] |
| HBMEC | Brain | *** | [69,94] |
| SNB19 | CNS in brain | *** | [42] |
| Huh-7 | Liver | *** | [70] |
| HFF-1 | Skin | *** | [64] |
| A549 | Lung | *** | [100,101] |
| HOBIT | Osteoblast-like Cells | *** | [102] |
| Cellular Antiviral Responses to Zika Infection | |||
| Cellular Response | Cellular Protein Involved | Molecular Actions and Consequences | References |
| Pro-inflammatory CD8+ T-cell immune response | Cytokines: IL-1β, IL-6, MIP1α; chemokines: IP-10, RANTES | T-cell mediated polyfunctional immune responses with releases of antiviral cytokines and chemokines | [65,103,118,119] |
| CD14+ monocytes and macrophages immune response | CXCL9, CXCL10, CXCL11, CCL5, IL-15 | CD14+ monocytes prime NK cell activities during ZIKV infection | [92] |
| Humoral immune response | IgM, IgG | Production of neutralizing and protective antibodies to ZIKV | [120,121,122] |
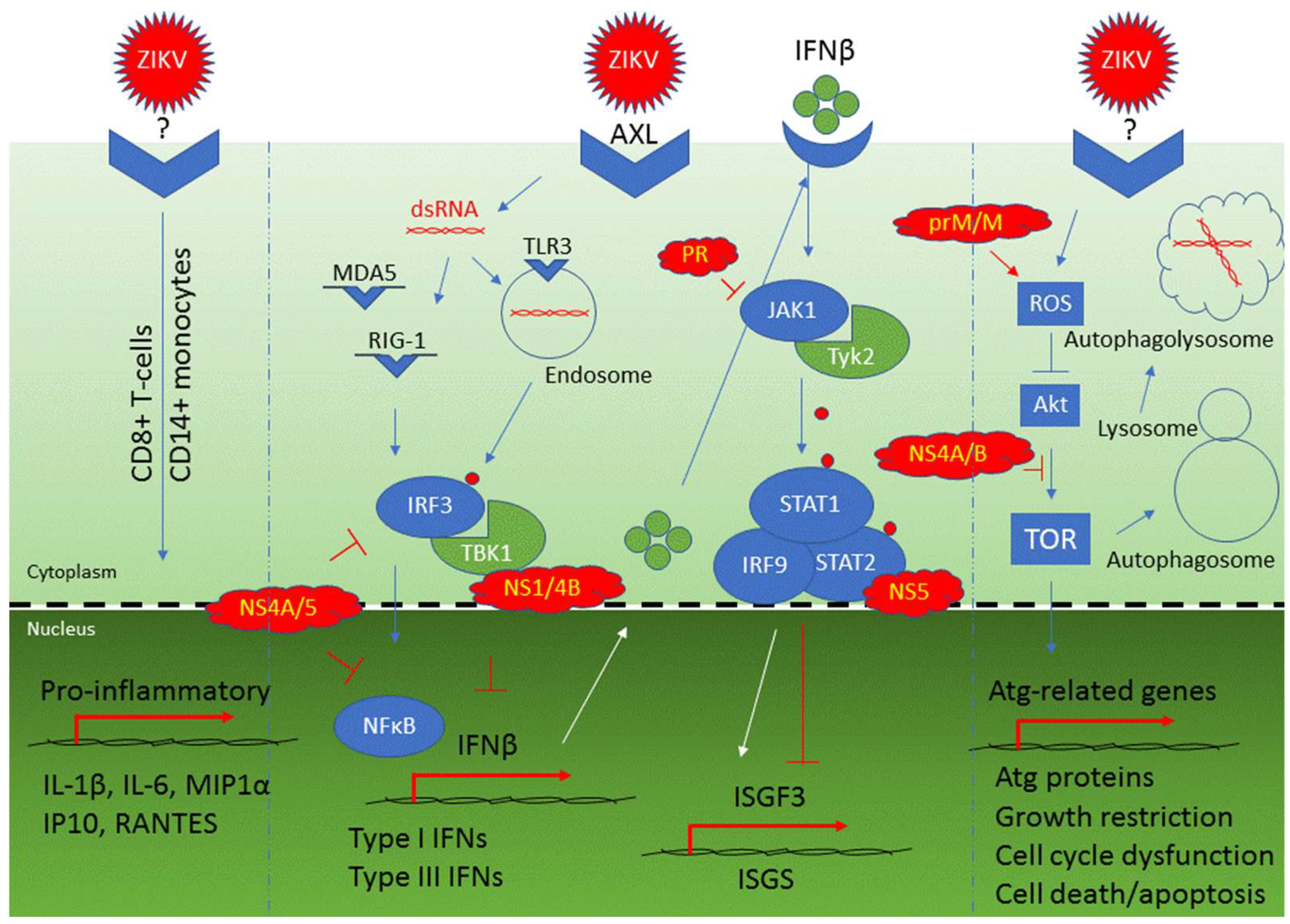
| Cellular innate immune response: TLR3-mediated response | TLR3, IRF3, TBK1, type I IFNs, and IFNβ | An early response that triggers IRF3 and recognizes ZIKV dsRNA in cytoplasm leading to activation of type I IFNs and IFNβ production | [57,64,65] |
| Cellular innate immune response: RIG-1/MDA5-mediated response | RIG-1, MDA5, IRF-3, NFkB, type I IFNs, and IFNβ | Late responses that recognize ZIKV dsRNA and contribute to activation of type I IFNs and IFNβ production | [64,65] |
| Type I and type III interferon activation | OAS2, ISG15, MX1 | Production of IFNβ as part of the cellular antiviral responses | [64] |
| Viral Counteraction | |||
| Viral response | Viral protein involved | Molecular actions and consequences | References |
| Counteraction to activation of type 1 IFNs and IFNβ production | NS1, NS2A, NS2B, NS4A, NS4B and NS5 | Targeting RIG-1 pathway | [123,124,125] |
| Inhibition of IFNβ production | NS1, NS4A, NS4B, NS5 | NS4A and NS5 inhibit IRF3 and NFkB; NS1 inhibits IRF3 IFNβ production through binding to TBK1 | [49] |
| Inhibition of the JAK/STAT pathway | NS5, PR | NS5 binds to STAT2 for its proteasomal degradation; PR inhibits JAK1 kinase | [115,126,127] |
| Selective activation of type II IFN signaling | NS5 | NS5 promotes the formation of STAT1/STAT1 homodimers and activates type II IFN for viral replication | [123] |
| Induction of cellular autophagy | prM, M, NS1, NS2A, NS4A | In a yeast study, these ZIKV proteins induced cellular autophagy as indicated by formation of cytoplasmic puncta | [9] |
| Induction of cellular autophagy | NS4A, NS4B | Inhibit Akt-mediated mTOR pathway through Tor1/TSC1 and Tip41 | [9,111] |
| Protein | Primary Function | Main Phenotypes | References |
|---|---|---|---|
| Structural Proteins | |||
| anaC | Anchored capsid protein | In the fission yeast cells, it restricts cellular growth and affects cell cycling. It also induces cellular oxidative stress leading to cell death. | [9] |
| C | Capsid protein | In the fission yeast cells, it restricts cellular growth. It also induces cellular oxidative stress leading to cell death; in hNPCs, it induces ribosomal stress and apoptosis. | [9,130] |
| prM | Precursor membrane protein | In the fission yeast cells, it restricts cellular growth and affects cell cycling. It also induces cellular oxidative stress and autophagy leading to cell death; a single prM mutation contributes to fetal microcephaly | [9,131] |
| M | Membrane protein | In the fission yeast cells, it restricts cellular growth and affects cell cycling. It also induces cellular oxidative stress and autophagy, leading to cell death. | [9] |
| Pr | Cleaved product from prM | Unknown | |
| E | Envelope protein | A putative cytopathic factor based on a yeast study. E protein facilitates viral entry. A single residue in the αB helix of the E protein is critical for Zika virus thermostability, and interaction with the host cell membrane. | [9,132] |
| Non-structural Proteins | |||
| NS1 | Viral replication, pathogenesis and immune evasion | In the fission yeast cells, it induces cellular oxidative stress and autophagy leading to cell death; An essential role in viral replication and immune evasion. It presents on the cell surface and presents as a dimer within cells, and as a hexamer once being secreted. NS1-mediated CPEs in mammalian cells have not yet been established. | [47,48,49,133] |
| NS2A | Unknown | In the fission yeast cells, it induces cellular oxidative stress and autophagy leading to cell death; ZIKV-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction (AJ) proteins, leading to reduced proliferation and premature differentiation of radial glial cells and aberrant positioning of newborn neurons. | [131] |
| NS2B | Protease cofactor | In fission yeast cells, it restricts cellular growth. Forms a protease complex with NS3; a putative cytopathic factor based on a yeast study | [9,134] |
| NS3 | Protease and helicase | NS3-mediated CPEs in mammalian cells have not yet been established. | [131] |
| NS4A | Viral RNA synthesis and viral morphogenesis | In the fission yeast cells, it restricts cellular growth and affects cell cycling. It also induces cellular oxidative stress and autophagy leading to cell death. It induces autophagy by inhibiting Atk-mediated TOR pathway through Tor1/TSC1 and Tip41 in both yeast and mammalian cells. | [9,111] |
| 2K | A signal peptide | Viral RNA synthesis and viral morphogenesis. 2K-mediated CPEs have not yet been established. | [9,11,12] |
| NS4B | Viral RNA synthesis and viral morphogenesis | Synergistic to NS4A on inhibiting Akt-mediated TOR pathway | [111] |
| NS5 | Methyltrasferase; RNA-dependent polymerase | NS5-mediated CPEs in mammalian cells have not yet been established. | [128] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, I.; Bos, S.; Li, G.; Wang, S.; Gadea, G.; Desprès, P.; Zhao, R.Y. Probing Molecular Insights into Zika Virus–Host Interactions. Viruses 2018, 10, 233. https://doi.org/10.3390/v10050233
Lee I, Bos S, Li G, Wang S, Gadea G, Desprès P, Zhao RY. Probing Molecular Insights into Zika Virus–Host Interactions. Viruses. 2018; 10(5):233. https://doi.org/10.3390/v10050233
Chicago/Turabian StyleLee, Ina, Sandra Bos, Ge Li, Shusheng Wang, Gilles Gadea, Philippe Desprès, and Richard Y. Zhao. 2018. "Probing Molecular Insights into Zika Virus–Host Interactions" Viruses 10, no. 5: 233. https://doi.org/10.3390/v10050233
APA StyleLee, I., Bos, S., Li, G., Wang, S., Gadea, G., Desprès, P., & Zhao, R. Y. (2018). Probing Molecular Insights into Zika Virus–Host Interactions. Viruses, 10(5), 233. https://doi.org/10.3390/v10050233