Following Acute Encephalitis, Semliki Forest Virus is Undetectable in the Brain by Infectivity Assays but Functional Virus RNA Capable of Generating Infectious Virus Persists for Life


Abstract
:
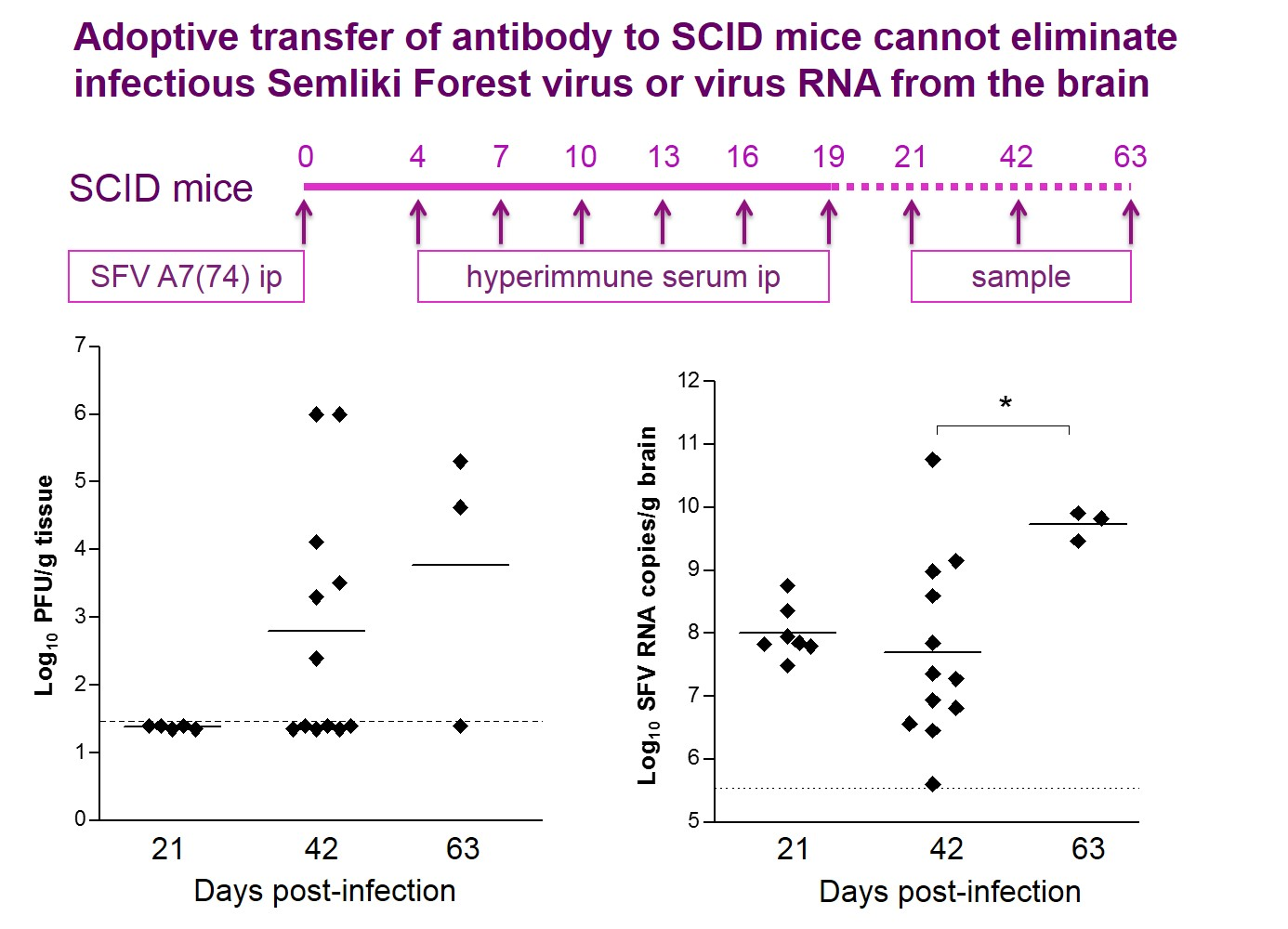{kind=link}
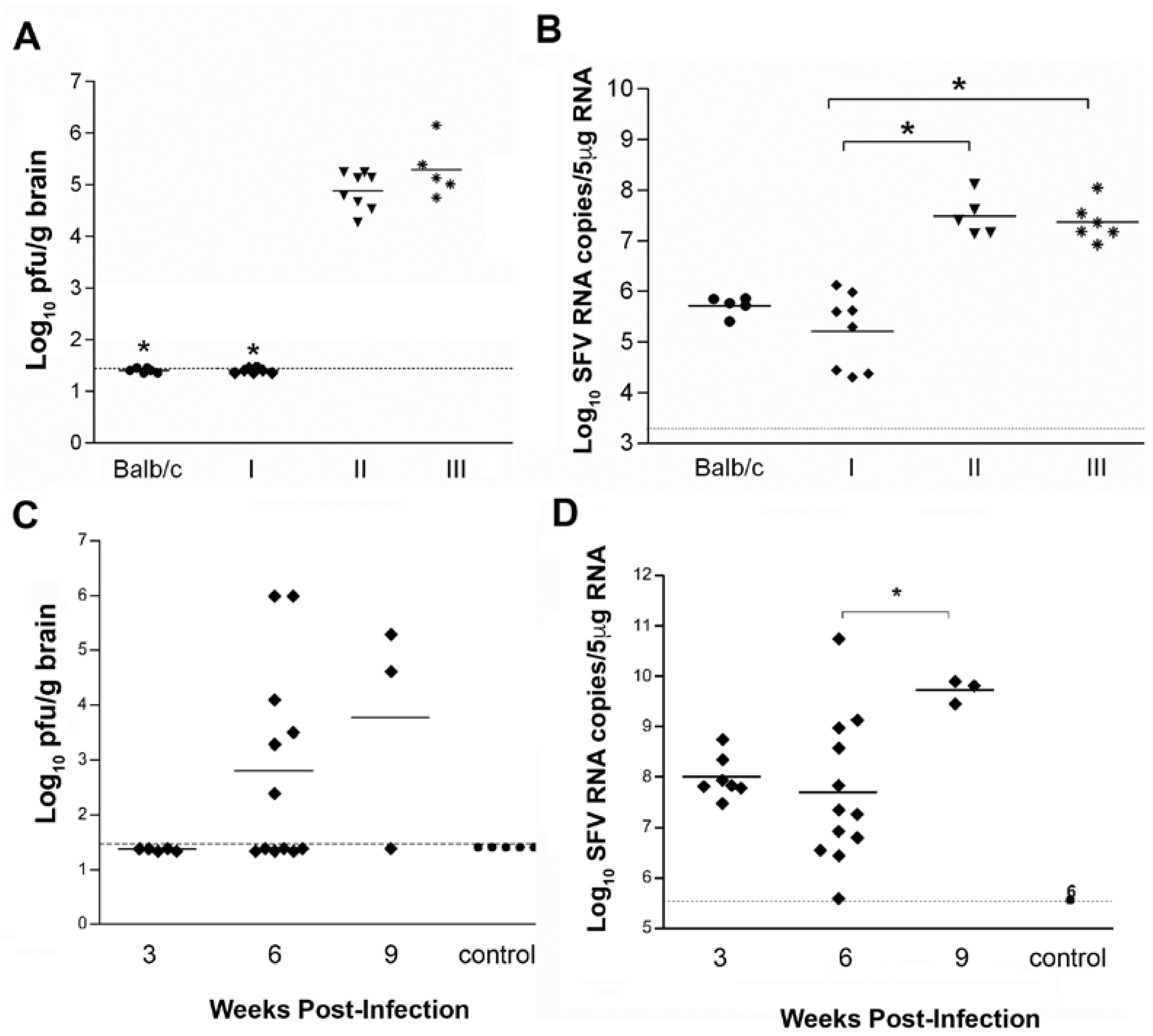{kind=link}
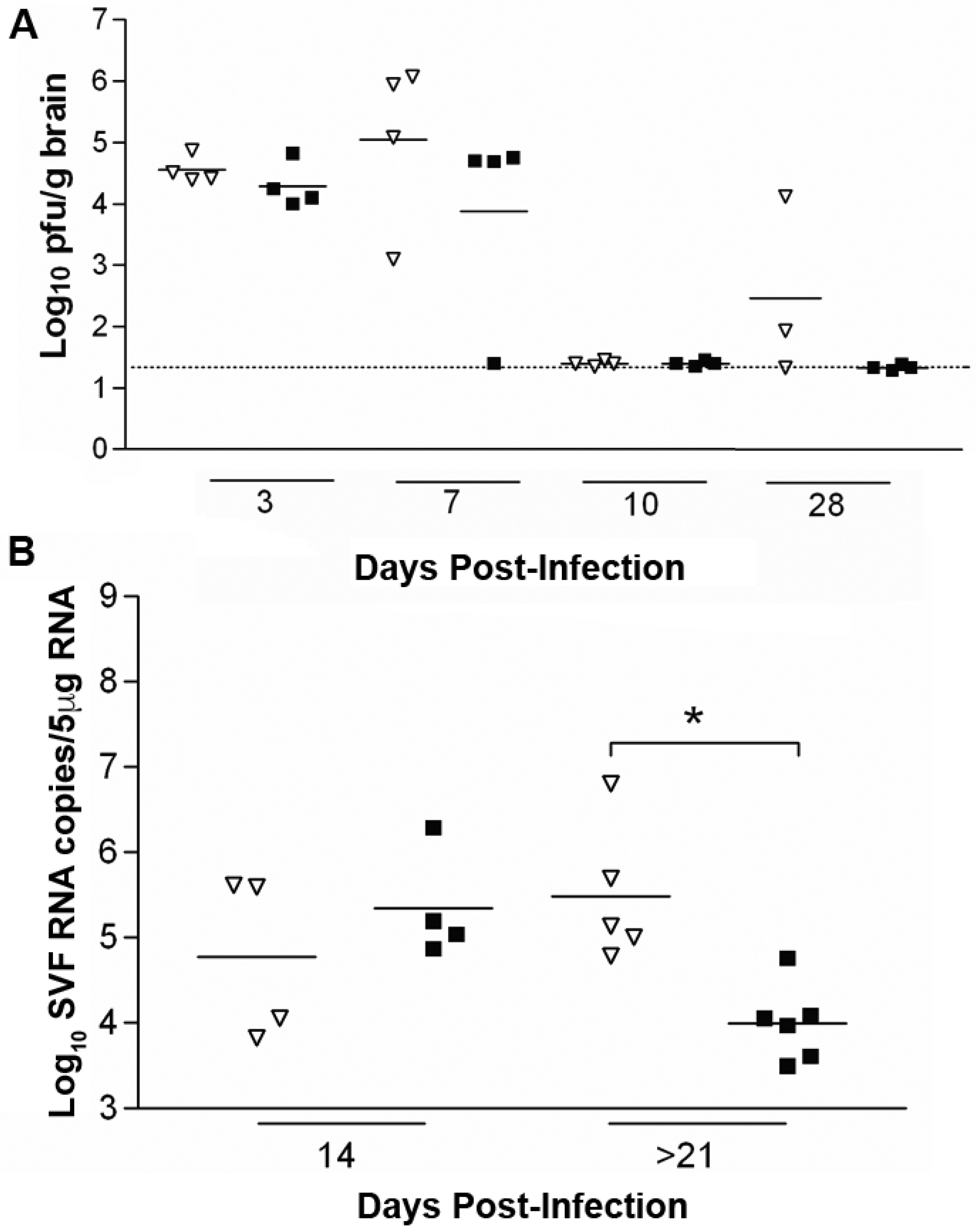{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Virus
2.2. Mice
2.3. Production of Hyperimmune (HI) Serum
2.4. RNA Extraction
2.5. Quantitative RT-PCR
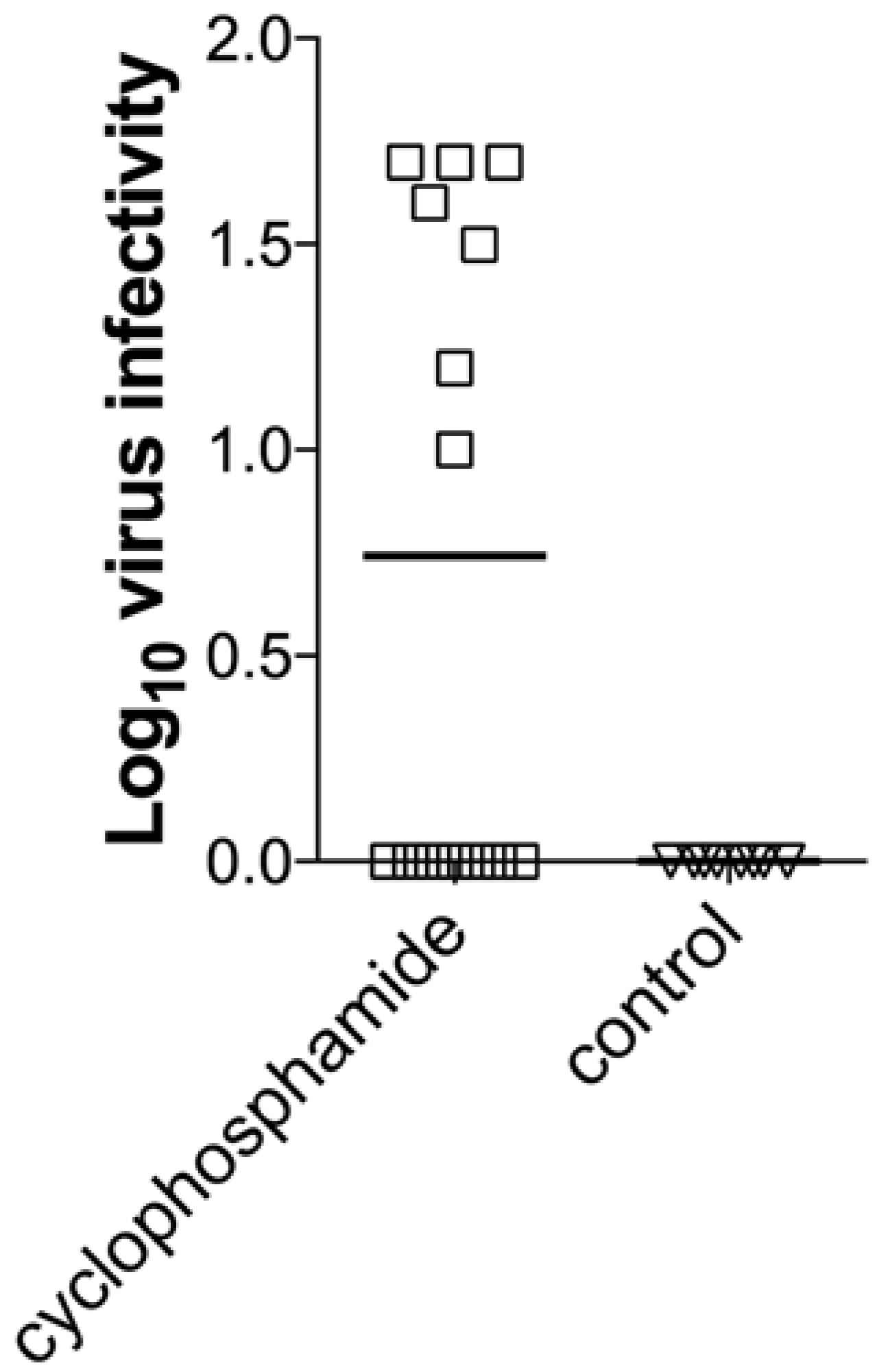
2.6. Cyclophosphamide Treatment
3. Results
3.1. Antibodies Are Sufficient to Reduce Infectious Virus in The Brain to Undetectable Levels but Cannot Eliminate All Potentially Infectious Virus Material
3.2. MHC Class II Knockout Mice Are Unable to Clear Infectious Virus from The Brain
3.3. Lack of CD8+ T Cells Leads to Delayed Viral RNA Clearance in The Infected Brain
3.4. In Immunocompetent Mice, Infectious Virus Can Also Be Recovered Many Months after Infection
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, A.; Lobner, M.; Cedile, O.; Owens, T. Comparison of microglia and infiltrating CD11c(+) cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflamm. 2014, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, H.; Schmidt, H.; Cavalie, A.; Jenne, D.; Wekerle, H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: Differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J. Exp. Med. 1997, 185, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Cebrian, C.; Loike, J.D.; Sulzer, D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson’s and other brain diseases. Front. Neuroanat. 2014, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.R.; Webb, J.; Rebus, S.; Walker, R.; Williams, A.; Fazakerley, J.K. Inducible cytokine gene expression in the brain in the ME7/CV mouse model of scrapie is highly restricted, is at a strikingly low level relative to the degree of gliosis and occurs only late in disease. J. Gen. Virol. 2003, 84 Pt 9, 2605–2611. [Google Scholar] [CrossRef] [PubMed]
- Wekerle, H.; Linington, C.; Lassmann, H.; Meyermann, R. Cellular immune reactivity within the CNS. Trends Neurosci. 1986, 9, 271–277. [Google Scholar] [CrossRef]
- Allsopp, T.E.; Fazakerley, J.K. Altruistic cell suicide and the specialized case of the virus-infected nervous system. Trends Neurosci. 2000, 23, 284–290. [Google Scholar] [CrossRef]
- Atkins, G.J.; Sheahan, B.J.; Liljestrom, P. The molecular pathogenesis of Semliki Forest virus: A model virus made useful? J. Gen. Virol 1999, 80 Pt 9, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, J.K. Pathogenesis of Semliki Forest virus encephalitis. J. Neurovirol. 2002, 8 (Suppl. 2), 66–74. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, J.K.; Pathak, S.; Scallan, M.; Amor, S.; Dyson, H. Replication of the A7(74) strain of Semliki Forest virus is restricted in neurons. Virology 1993, 195, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, J.K.; Cotterill, C.L.; Lee, G.; Graham, A. Virus tropism, distribution, persistence and pathology in the corpus callosum of the Semliki Forest virus-infected mouse brain: A novel system to study virus-oligodendrocyte interactions. Neuropathol. Appl. Neurobiol. 2006, 32, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Fragkoudis, R.; Tamberg, N.; Siu, R.; Kiiver, K.; Kohl, A.; Merits, A.; Fazakerley, J.K. Neurons and oligodendrocytes in the mouse brain differ in their ability to replicate Semliki Forest virus. J. NeuroVirol. 2009, 15, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Subak-Sharpe, I.; Dyson, H.; Fazakerley, J. In vivo depletion of CD8+ T cells prevents lesions of demyelination in Semliki Forest virus infection. J. Virol. 1993, 67, 7629–7633. [Google Scholar] [PubMed]
- Fragkoudis, R.; Ballany, C.M.; Boyd, A.; Fazakerley, J.K. In Semliki Forest virus encephalitis, antibody rapidly clears infectious virus and is required to eliminate viral material from the brain, but is not required to generate lesions of demyelination. J. Gen. Virol. 2008, 89 Pt 10, 2565–2568. [Google Scholar] [CrossRef] [PubMed]
- Parsons, L.M.; Webb, H.E. Virus titres and persistently raised white cell counts in cerebrospinal fluid in mice after peripheral infection with demyelinating Semliki Forest virus. Neuropathol. Appl. Neurobiol. 1982, 8, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Parsons, L.M.; Webb, H.E. Identification of immunoglobulin-containing cells in the central nervous system of the mouse following infection with the demyelinating strain of Semliki Forest virus. Br. J. Exp. Pathol. 1989, 70, 247–255. [Google Scholar] [PubMed]
- Levine, B.; Griffin, D.E. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J. Virol. 1992, 66, 6429–6435. [Google Scholar] [PubMed]
- Tyor, W.R.; Wesselingh, S.; Levine, B.; Griffin, D.E. Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J. Immunol. 1992, 149, 4016–4020. [Google Scholar] [PubMed]
- Fazakerley, J.K.; Webb, H.E. Semliki Forest virus-induced, immune-mediated demyelination: Adoptive transfer studies and viral persistence in nude mice. J. Gen. Virol. 1987, 68 Pt 2, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Scallan, M.F.; Morris, M.M.; Dyson, H.; Fazakerley, J.K. Role of immune responses in protection and pathogenesis during Semliki Forest virus encephalitis. J. Gen. Virol. 1996, 77 Pt 2, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Hardwick, J.M.; Trapp, B.D.; Crawford, T.O.; Bollinger, R.C.; Griffin, D.E. Antibody-mediated clearance of alphavirus infection from neurons. Science 1991, 254, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.; Gray, D.; Dierich, A.; Kaufman, J.; Lemeur, M.; Benoist, C.; Mathis, D. Mice lacking MHC class II molecules. Cell 1991, 66, 1051–1066. [Google Scholar] [CrossRef]
- Fung-Leung, W.P.; Schilham, M.W.; Rahemtulla, A.; Kundig, T.M.; Vollenweider, M.; Potter, J.; van Ewijk, W.; Mak, T.W. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell 1991, 65, 443–449. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003, 3, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, J.K.; Southern, P.; Bloom, F.; Buchmeier, M.J. High resolution in situ hybridization to determine the cellular distribution of lymphocytic choriomeningitis virus RNA in the tissues of persistently infected mice: Relevance to arenavirus disease and mechanisms of viral persistence. J. Gen. Virol. 1991, 72 Pt 7, 1611–1625. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, J.O.; Jacobson, S. Viruses and multiple sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 528–544. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.; Leparc-Goffart, I.; Lina, B.; Fuchs, F.; Foray, S.; Janatova, I.; Aymard, M.; Kopecka, H. Postpolio syndrome: Poliovirus persistence is involved in the pathogenesis. J. Neurol. 1999, 246, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.; Bonthius, D.J. Measles virus and associated central nervous system sequelae. Semin. Pediatr. Neurol. 2012, 19, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Allsopp, T.E.; Scallan, M.F.; Williams, A.; Fazakerley, J.K. Virus infection induces neuronal apoptosis: A comparison with trophic factor withdrawal. Cell Death Differ. 1998, 5, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, J.K.; Allsopp, T.E. Programmed cell death in virus infections of the nervous system. Curr. Top. Microbiol. Immunol. 2001, 253, 95–119. [Google Scholar] [PubMed]
- Donnelly, S.M.; Sheahan, B.J.; Atkins, G.J. Long-term effects of Semliki Forest virus infection in the mouse central nervous system. Neuropathol. Appl. Neurobiol. 1997, 23, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, A.P.; Durbin, J.E.; Griffin, D.E. Control of Sindbis virus infection by antibody in interferon-deficient mice. J. Virol. 2000, 74, 3905–3908. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E.; Johnson, R.T. Role of the immune response in recovery from Sindbis virus encephalitis in mice. J. Immunol. 1977, 118, 1070–1075. [Google Scholar] [PubMed]
- Hirsch, R.L.; Griffin, D.E.; Johnson, R.T. Interactions between immune cells and antibody in protection from fatal Sindbis virus encephalitis. Infect. Immun. 1979, 23, 320–324. [Google Scholar] [PubMed]
- Rowell, J.F.; Griffin, D.E. Contribution of T cells to mortality in neurovirulent Sindbis virus encephalomyelitis. J. Neuroimmunol. 2002, 127, 106–114. [Google Scholar] [CrossRef]
- Brooke, C.B.; Deming, D.J.; Whitmore, A.C.; White, L.J.; Johnston, R.E. T cells facilitate recovery from Venezuelan equine encephalitis virus-induced encephalomyelitis in the absence of antibody. J. Virol. 2010, 84, 4556–4568. [Google Scholar] [CrossRef] [PubMed]
- Burdeinick-Kerr, R.; Griffin, D.E. Gamma interferon-dependent, noncytolytic clearance of sindbis virus infection from neurons in vitro. J. Virol. 2005, 79, 5374–5385. [Google Scholar] [CrossRef] [PubMed]
- Burdeinick-Kerr, R.; Wind, J.; Griffin, D.E. Synergistic roles of antibody and interferon in noncytolytic clearance of Sindbis virus from different regions of the central nervous system. J. Virol. 2007, 81, 5628–5636. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Griffin, D.E. The role of CD8(+) T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J. Virol. 2000, 74, 6117–6125. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.D.; Pavelko, K.D.; Leibowitz, J.; Lin, X.; Rodriguez, M. CD4(+) and CD8(+) T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J. Virol. 1998, 72, 7320–7329. [Google Scholar] [PubMed]
- Lane, T.E.; Liu, M.T.; Chen, B.P.; Asensio, V.C.; Samawi, R.M.; Paoletti, A.D.; Campbell, I.L.; Kunkel, S.L.; Fox, H.S.; Buchmeier, M.J. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 2000, 74, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.R.; Kongsgaard, M.; Steffensen, M.A.; Fenger, C.; Rasmussen, M.; Skjodt, K.; Finsen, B.; Stryhn, A.; Buus, S.; Christensen, J.P.; et al. CD8+ T cells complement antibodies in protecting against yellow fever virus. J. Immunol. 2015, 194, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Arora, K.L.; Rawat, S.; Chaturvedi, U.C. Persistence, latency and reactivation of Japanese encephalitis virus infection in mice. J. Gen. Virol. 1986, 67 Pt 2, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Appler, K.K.; Brown, A.N.; Stewart, B.S.; Behr, M.J.; Demarest, V.L.; Wong, S.J.; Bernard, K.A. Persistence of West Nile virus in the central nervous system and periphery of mice. PLoS ONE 2010, 5, e10649. [Google Scholar] [CrossRef] [PubMed]
- Gea-Banacloche, J.C. Rituximab-associated infections. Semin. Hematol. 2010, 47, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.H.; Ciarlini, P.; Santagata, S.; Ahmed, A.A.; De Girolami, U.; Prasad, S.; Mukerji, S.S. Fatal Eastern Equine Encephalitis in a Patient on Maintenance Rituximab: A Case Report. Open Forum Infect. Dis. 2017, 4, ofx021. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fragkoudis, R.; Dixon-Ballany, C.M.; Zagrajek, A.K.; Kedzierski, L.; Fazakerley, J.K. Following Acute Encephalitis, Semliki Forest Virus is Undetectable in the Brain by Infectivity Assays but Functional Virus RNA Capable of Generating Infectious Virus Persists for Life. Viruses 2018, 10, 273. https://doi.org/10.3390/v10050273
Fragkoudis R, Dixon-Ballany CM, Zagrajek AK, Kedzierski L, Fazakerley JK. Following Acute Encephalitis, Semliki Forest Virus is Undetectable in the Brain by Infectivity Assays but Functional Virus RNA Capable of Generating Infectious Virus Persists for Life. Viruses. 2018; 10(5):273. https://doi.org/10.3390/v10050273
Chicago/Turabian StyleFragkoudis, Rennos, Catherine M. Dixon-Ballany, Adrian K. Zagrajek, Lukasz Kedzierski, and John K. Fazakerley. 2018. "Following Acute Encephalitis, Semliki Forest Virus is Undetectable in the Brain by Infectivity Assays but Functional Virus RNA Capable of Generating Infectious Virus Persists for Life" Viruses 10, no. 5: 273. https://doi.org/10.3390/v10050273
APA StyleFragkoudis, R., Dixon-Ballany, C. M., Zagrajek, A. K., Kedzierski, L., & Fazakerley, J. K. (2018). Following Acute Encephalitis, Semliki Forest Virus is Undetectable in the Brain by Infectivity Assays but Functional Virus RNA Capable of Generating Infectious Virus Persists for Life. Viruses, 10(5), 273. https://doi.org/10.3390/v10050273