Whole Genome Analysis of Two Novel Type 2 Porcine Reproductive and Respiratory Syndrome Viruses with Complex Genome Recombination between Lineage 8, 3, and 1 Strains Identified in Southwestern China



Abstract
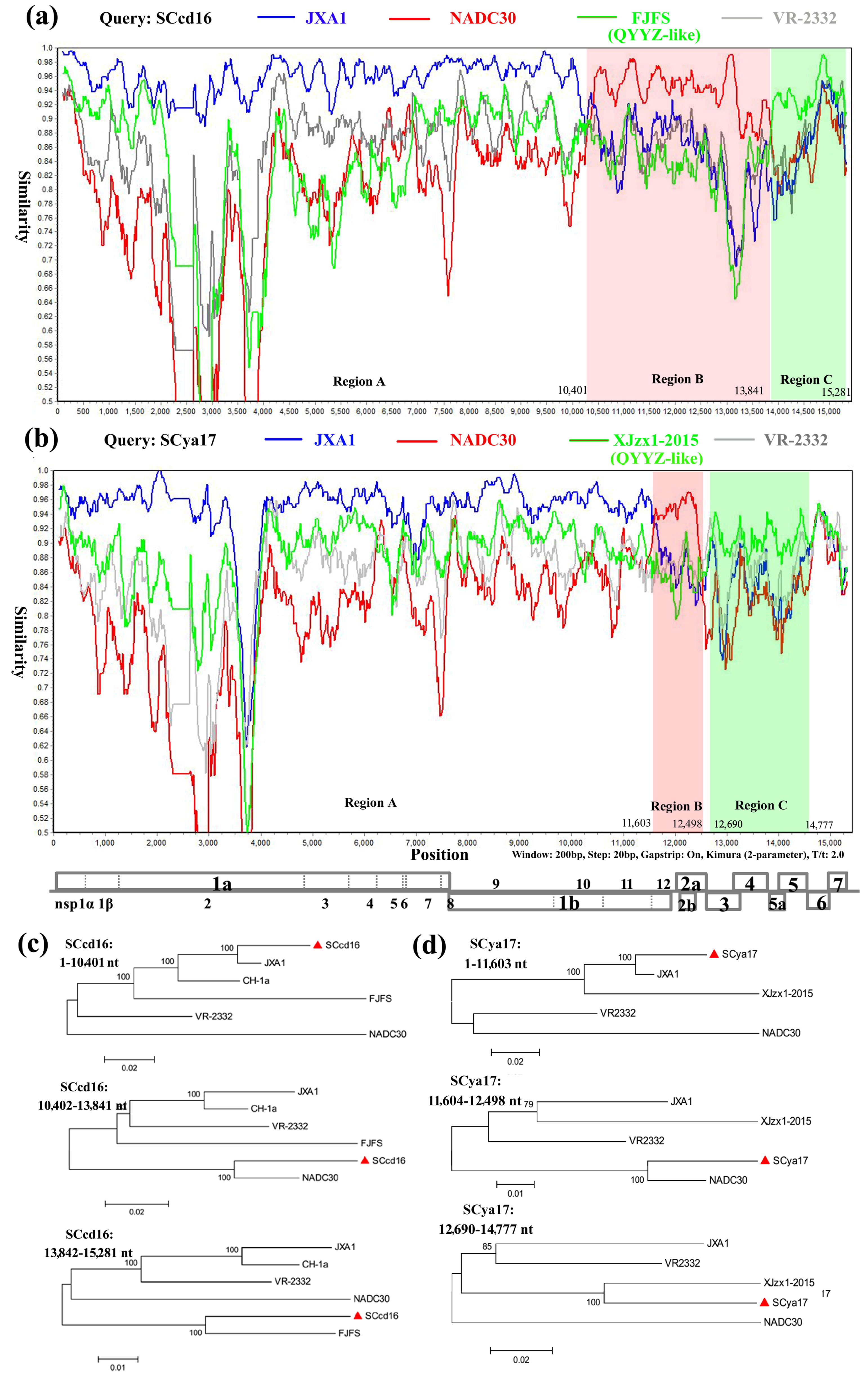
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Virus Isolation
2.3. Viral Genome Extraction, RACE and RT-PCR
2.4. Genome Alignment and Phylogenetic Analysis
2.5. Recombinant Analysis
3. Results
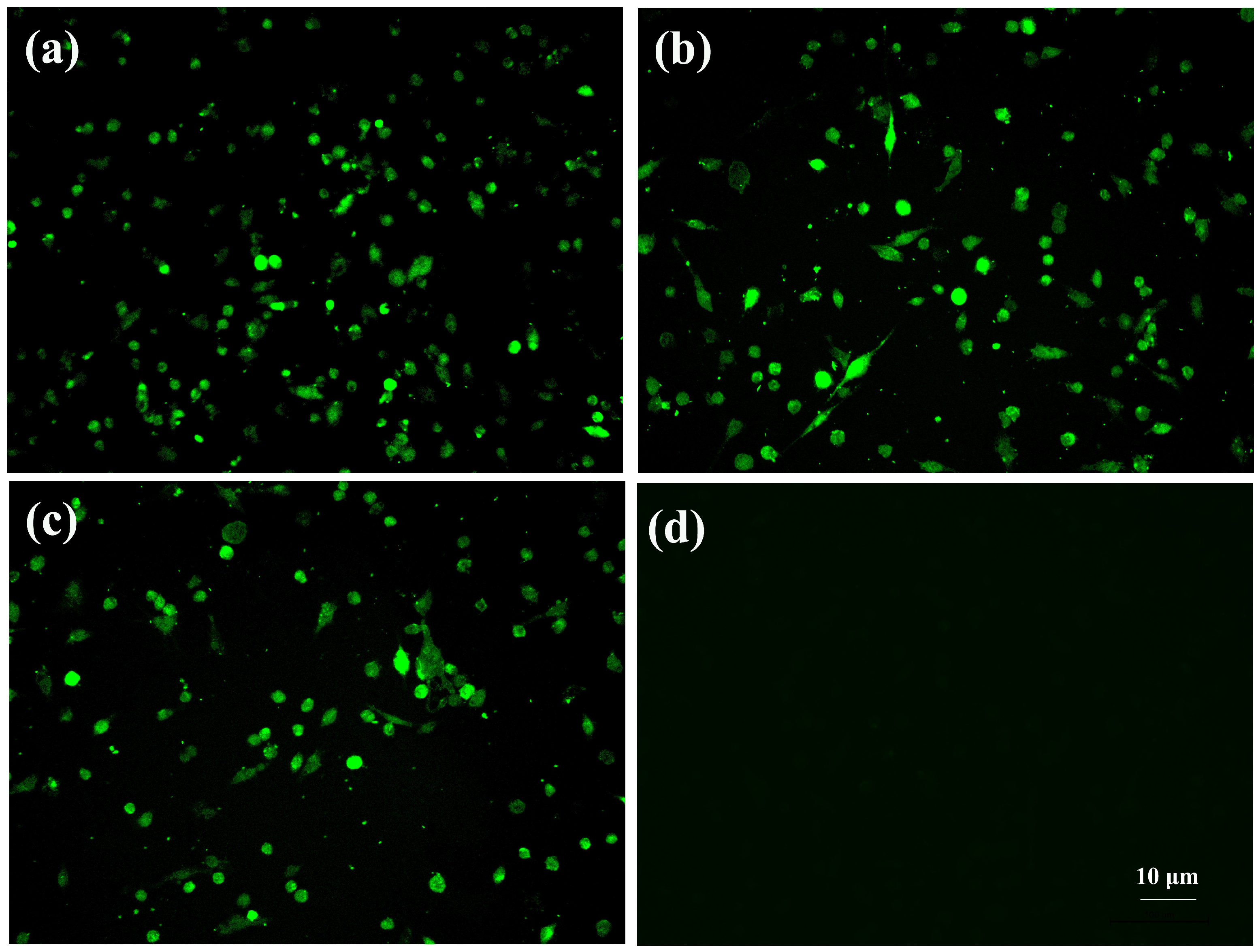
3.1. Virus Isolation
3.2. Genomic Characterization and Homology Analysis
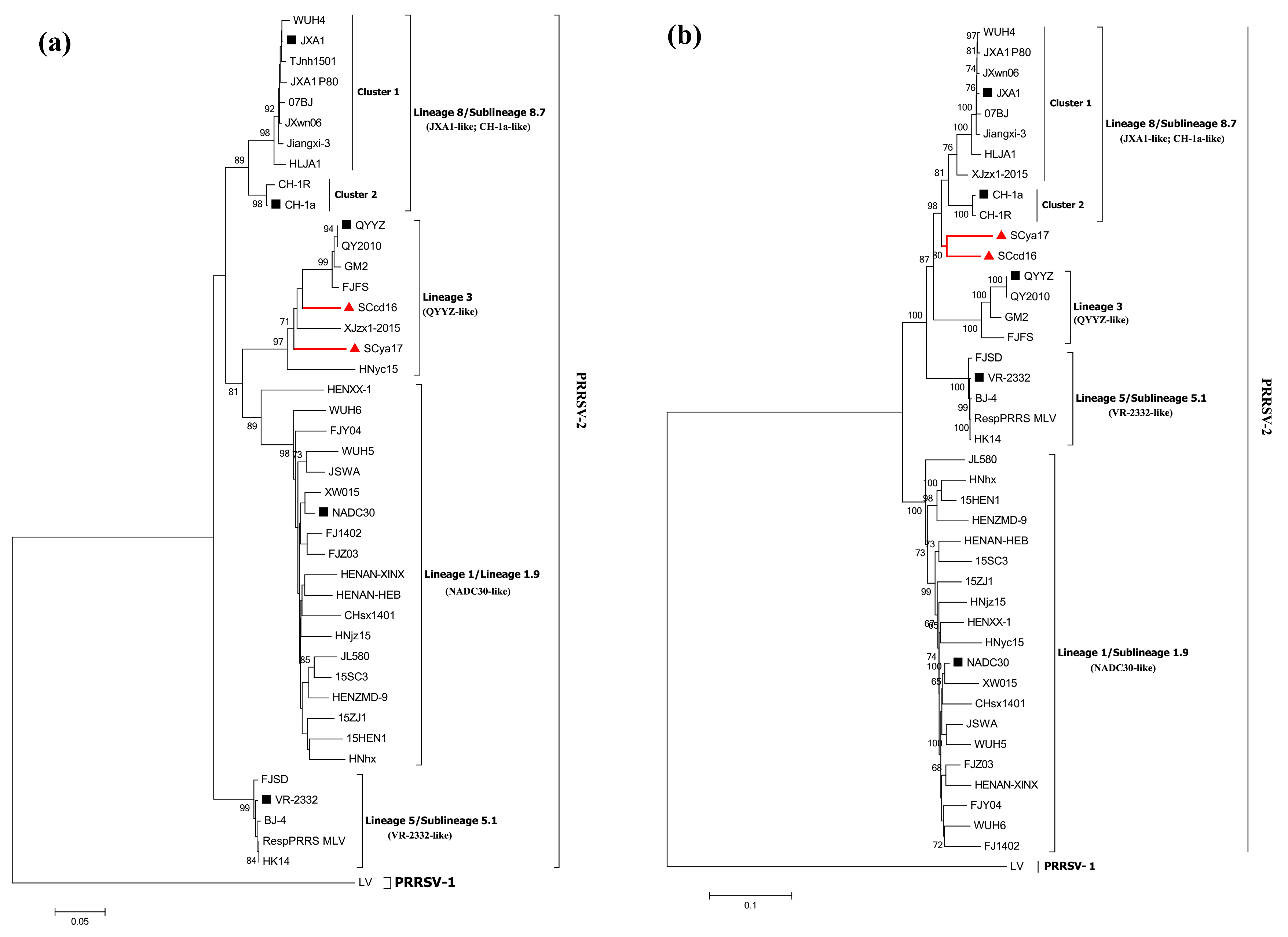
3.3. Phylogenetic Analysis
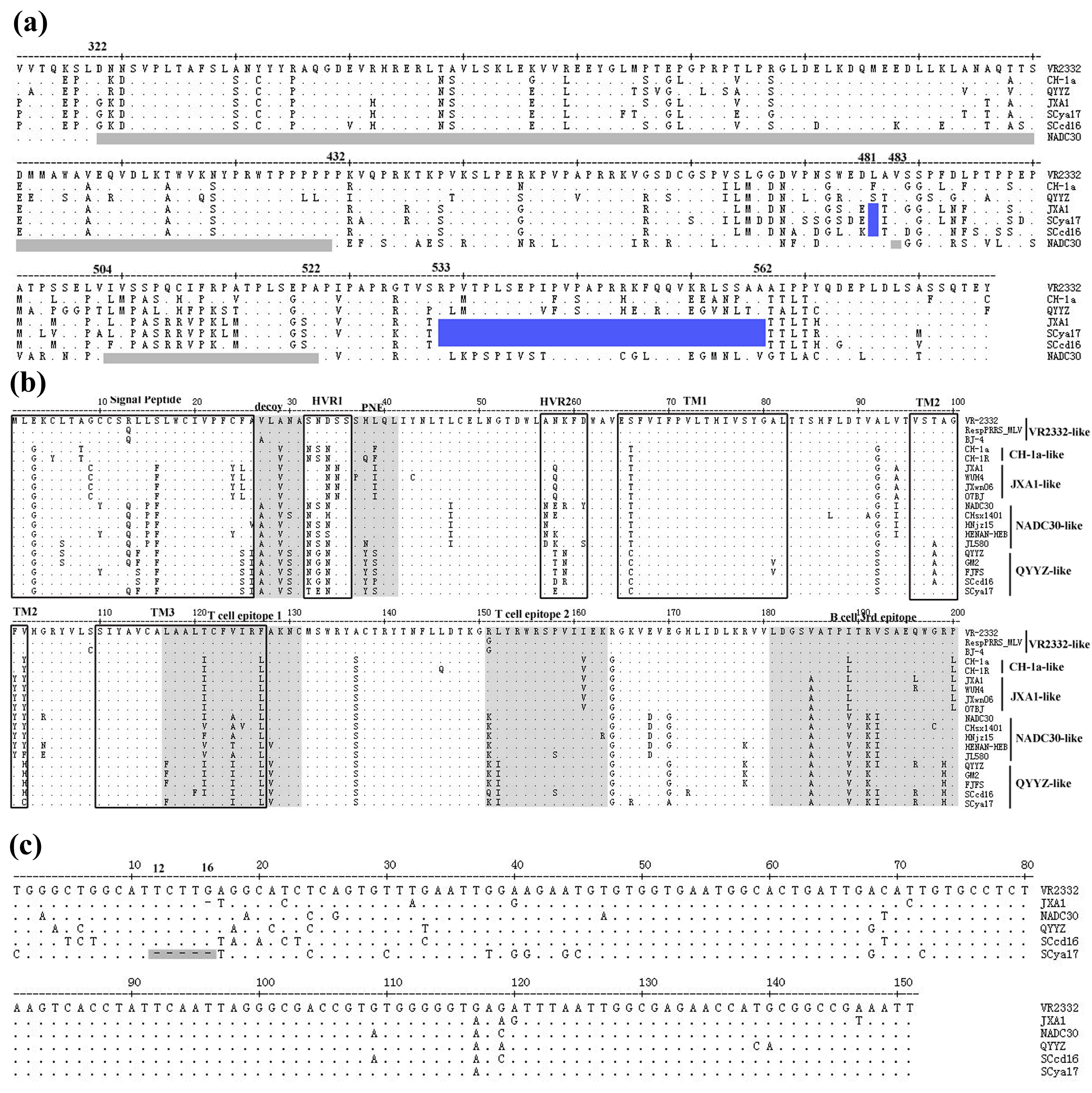
3.4. Sequence Analysis of Nsp2, GP5 and 3′-UTR
3.5. Recombination Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Keffaber, K. Reproductive failure of unknown etiol-ogy. Am. Assoc. Swine Pract. Newsl. 1989, 1, 1–9. [Google Scholar]
- Wensvoort, G.; Terpstra, C.; Pol, J.M.; ter Laak, E.A.; Bloemraad, M.; de Kluyver, E.P.; Kragten, C.; van Buiten, L.; den Besten, A.; Wagenaar, F.; et al. Mystery swine disease in the Netherlands: The isolation of lelystad virus. Vet. Q. 1991, 13, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, D. Nidovirales: A new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 1997, 142, 629–633. [Google Scholar] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef] [PubMed]
- Meulenberg, J.J.; Petersen-den Besten, A.; De Kluyver, E.P.; Moormann, R.J.; Schaaper, W.M.; Wensvoort, G. Characterization of proteins encoded by ORFs 2 to 7 of lelystad virus. Virology 1995, 206, 155–163. [Google Scholar] [CrossRef]
- Murtaugh, M.P.; Elam, M.R.; Kakach, L.T. Comparison of the structural protein coding sequences of the VR-2332 and lelystad virus strains of the PRRSV virus. Arch. Virol. 1995, 140, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Van Dinten, L.C.; Wassenaar, A.L.; Gorbalenya, A.E.; Spaan, W.J.; Snijder, E.J. Processing of the equine arteritis virus replicase ORF1b protein: Identification of cleavage products containing the putative viral polymerase and helicase domains. J. Virol. 1996, 70, 6625–6633. [Google Scholar] [PubMed]
- Nelson, E.A.; Christopher-Hennings, J.; Drew, T.; Wensvoort, G.; Collins, J.E.; Benfield, D.A. Differentiation of U.S. and European isolates of porcine reproductive and respiratory syndrome virus by monoclonal antibodies. J. Clin. Microbiol. 1993, 31, 3184–3189. [Google Scholar] [PubMed]
- Guo, B.; Chen, Z.; Liu, W. Isolation and identification of porcine reproductory and respiratory syndrome (PRRSV) virus from aborted fetuses suspected of PRRS. Chin. J. Prev. Vet. Med. 1996, 2, 1–5. [Google Scholar]
- Shi, M.; Lam, T.T.; Hon, C.C.; Hui, R.K.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C. Molecular epidemiology of PRRSV: A phylogenetic perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lam, T.T.; Hon, C.C.; Murtaugh, M.P.; Davies, P.R.; Hui, R.K.; Li, J.; Wong, L.T.; Yip, C.W.; Jiang, J.W.; et al. Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J. Virol. 2010, 84, 8700–8711. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, X.X.; Li, R.; Qiao, S.; Zhang, G. The prevalent status and genetic diversity of porcine reproductive and respiratory syndrome virus in China: A molecular epidemiological perspective. Virol. J. 2018, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Yu, X.; Zhao, T.; Feng, Y.; Cao, Z.; Wang, C.; Hu, Y.; Chen, X.; Hu, D.; Tian, X.; et al. Emergence of fatal PRRSV variants: Unparalleled outbreaks of atypical PRRSV in China and molecular dissection of the unique hallmark. PLoS ONE 2007, 2, e526. [Google Scholar] [CrossRef] [PubMed]
- An, T.Q.; Zhou, Y.J.; Liu, G.Q.; Tian, Z.J.; Li, J.; Qiu, H.J.; Tong, G.Z. Genetic diversity and phylogenetic analysis of glycoprotein 5 of PRRSV isolates in mainland China from 1996 to 2006: Coexistence of two na-subgenotypes with great diversity. Vet. Microbiol. 2007, 123, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.H.; Tun, H.M.; Sun, B.L.; Mo, J.; Zhou, Q.F.; Deng, Y.X.; Xie, Q.M.; Bi, Y.Z.; Leung, F.C.; Ma, J.Y. Re-emerging of porcine respiratory and reproductive syndrome virus (lineage 3) and increased pathogenicity after genomic recombination with vaccine variant. Vet. Microbiol. 2015, 175, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, Z.; Ding, Y.; Ge, X.; Guo, X.; Yang, H. NADC30-like strain of porcine reproductive and respiratory syndrome virus, China. Emerg. Infect. Dis. 2015, 21, 2256–2257. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhuang, J.; Wang, J.; Han, L.; Sun, Z.; Xiao, Y.; Ji, G.; Li, Y.; Tan, F.; Li, X.; et al. Outbreak investigation of NADC30-like PRRSV in south-east China. Transbound. Emerg. Dis. 2016, 63, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Ye, C.; Chang, X.B.; Jiang, C.G.; Wang, S.J.; Cai, X.H.; Tong, G.Z.; Tian, Z.J.; Shi, M.; An, T.Q. Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J. Virol. 2015, 89, 10712–10716. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bao, H.; Wang, Y.; Tian, K. Widespread of NADC30-like PRRSV in China: Another pandora’s box for chinese pig industry as the outbreak of highly pathogenic PRRSV in 2006? Infect. Genet. Evol. 2006, 49, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; Zhou, X.; Zhai, J.Q.; Li, B.; Wei, C.H.; Dai, A.L.; Yang, X.Y.; Luo, M.L. Emergence of a novel highly pathogenic porcine reproductive and respiratory syndrome virus in China. Transbound. Emerg. Dis. 2017, 64, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Han, Q.; Zhang, L.; Zhang, Z.; Wu, Y.; Shen, H.; Jiang, P. Emergence of mosaic recombinant strains potentially associated with vaccine JXA1-R and predominant circulating strains of porcine reproductive and respiratory syndrome virus in different provinces of China. Virol. J. 2017, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, J.; Tan, F.; Li, Y.; Ji, G.; Zhuang, J.; Zhai, X.; Tian, K. Genome characterization of two NADC30-like porcine reproductive and respiratory syndrome viruses in China. SpringerPlus 2016, 5, 1677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, P.; Song, Z.; Lv, L.; Li, L.; Bai, J. Pathogenicity and antigenicity of a novel NADC30-like strain of porcine reproductive and respiratory syndrome virus emerged in China. Vet. Microbiol. 2016, 197, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Wan, B.; Guo, Z.; Qiao, S.; Li, R.; Xie, S.; Chen, X.X.; Zhang, G. Genomic analysis of a recombinant NADC30-like porcine reproductive and respiratory syndrome virus in China. Virus Genes 2018, 54, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bai, J.; Hou, H.; Song, Z.; Zhao, Y.; Jiang, P. A novel recombinant porcine reproductive and respiratory syndrome virus with significant variation in cell adaption and pathogenicity. Vet. Microbiol. 2017, 208, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.G.; Yu, L.Y.; Wang, P.P.; Zhang, L.Y.; Liu, Y.L.; Liang, P.S.; Song, C.X. A new recombined porcine reproductive and respiratory syndrome virus virulent strain in China. J. Vet. Sci. 2018, 19, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, J.; Yang, J.; Zeng, H.; Guo, L.; Ren, S.; Sun, W.; Chen, Z.; Cong, X.; Shi, J.; et al. Emergence of different recombinant porcine reproductive and respiratory syndrome viruses, China. Sci. Rep. 2018, 8, 4118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Kang, R.; Xie, B.; Tian, Y.; Wu, X.; Lv, X.; Yang, X.; Wang, H. Identification of a novel recombinant type 2 porcine reproductive and respiratory syndrome virus in China. Viruses 2018, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Kang, R.; Ji, G.; Tian, Y.; Ge, M.; Xie, B.; Yang, X.; Wang, H. Molecular characterization and recombination analysis of porcine reproductive and respiratory syndrome virus emerged in southwestern China during 2012–2016. Virus Genes 2018, 54, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, X.; Tian, Y.; Yin, S.; Geng, G.; Ge, X.; Guo, X.; Yang, H. Genetic diversity analysis of genotype 2 porcine reproductive and respiratory syndrome viruses emerging in recent years in China. BioMed Res. Int. 2014, 2014, 748068. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, H. Porcine reproductive and respiratory syndrome in China. Virus Res. 2010, 154, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fang, L.; Guo, X.; Gao, J.; Song, T.; Bi, J.; He, K.; Chen, H.; Xiao, S. Epidemiology and evolutionary characteristics of the porcine reproductive and respiratory syndrome virus in China between 2006 and 2010. J. Clin. Microbiol. 2011, 49, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; Wei, C.H.; Yang, X.Y.; Hou, X.L.; Dai, A.L.; Li, X.H.; Wei, M.K.; Pan, X.Z. Genetic diversity and evolutionary characterization of chinese porcine reproductive and respiratory syndrome viruses based on Nsp2 and ORF5. Arch. Virol. 2013, 158, 1811–1816. [Google Scholar] [CrossRef] [PubMed]
- Murtaugh, M.P.; Stadejek, T.; Abrahante, J.E.; Lam, T.T.; Leung, F.C. The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Kappes, M.A.; Faaberg, K.S. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 2015, 479, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Tian, K. NADC30-like porcine reproductive and respiratory syndrome in China. Open Virol. J. 2017, 11, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Dokland, T. The structural biology of PRRSV. Virus Res. 2010, 154, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, C.; Tan, F.; Gao, F.; Liu, P.; Qin, A.; Yuan, S. Identification of dispensable nucleotide sequence in 3′ untranslated region of porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ni, J.; Cao, Z.; Han, X.; Xia, Y.; Zi, Z.; Ning, K.; Liu, Q.; Cai, L.; Qiu, P.; et al. The epidemic status and genetic diversity of 14 highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) isolates from China in 2009. Vet. Microbiol. 2011, 150, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Sun, B.; Mo, J.; Zeng, X.; Zhang, G.; Wang, L.; Zhou, Q.; Zhu, L.; Li, Z.; Xie, Q.; et al. Attenuation and immunogenicity of a live high pathogenic PRRSV vaccine candidate with a 32-amino acid deletion in the NSP2 protein. J. Immunol. Res. 2014, 2014, 810523. [Google Scholar] [CrossRef] [PubMed]
- Brockmeier, S.L.; Loving, C.L.; Vorwald, A.C.; Kehrli, M.E., Jr.; Baker, R.B.; Nicholson, T.L.; Lager, K.M.; Miller, L.C.; Faaberg, K.S. Genomic sequence and virulence comparison of four type 2 porcine reproductive and respiratory syndrome virus strains. Virus Res. 2012, 169, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, Y.; Yu, L.; Zhang, P.; Liu, X.; Zhang, L.; Liu, Y.; Liang, P.; Wang, L.; Song, C. Pathogenicity of a newly emerged recombined porcine reproductive and respiratory syndrome virus strain (subgenotype III) in China. Vet. Microbiol. 2017, 210, 162–166. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
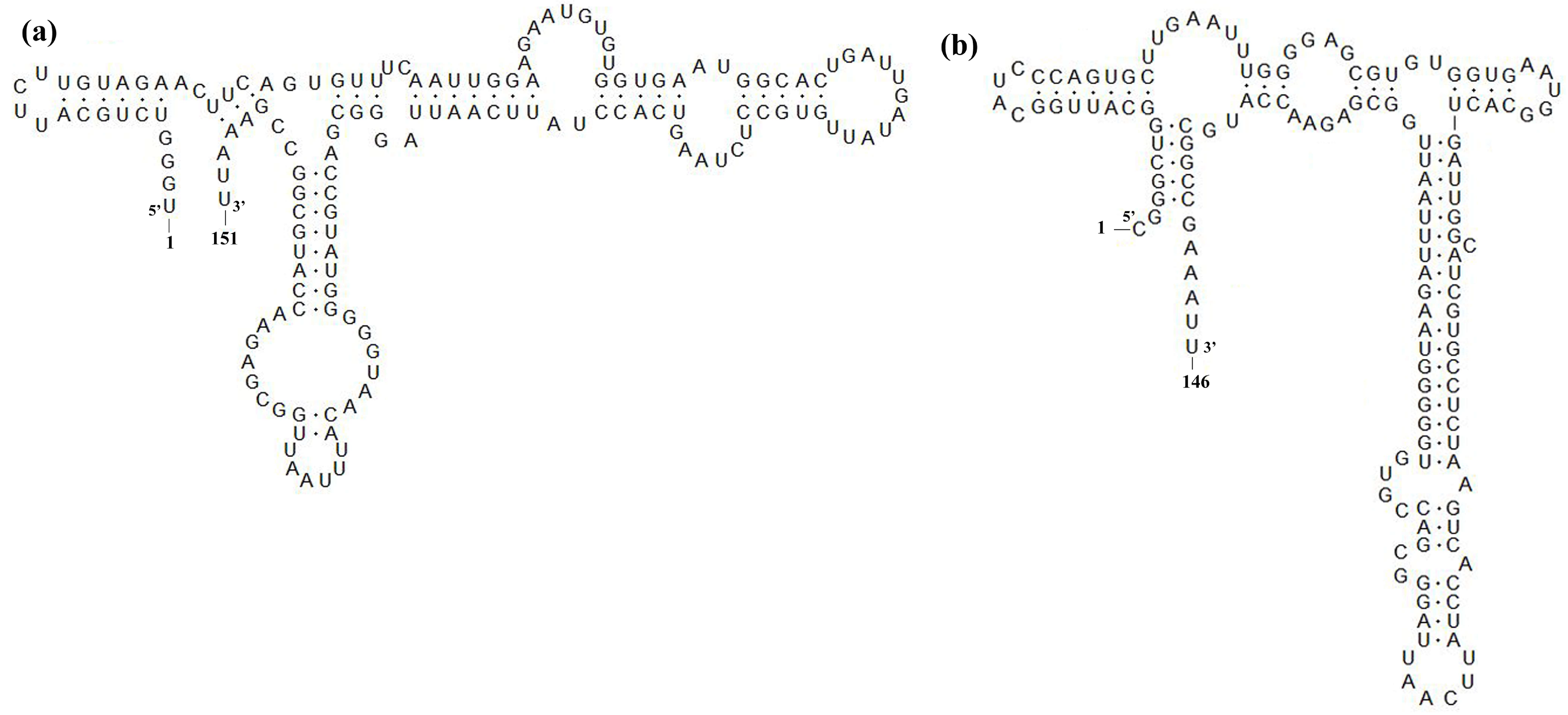{kind=link}
{kind=link}
| Region | Length (nt/aa) | Pairwise % Identity (nt/aa) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| JXA1 | VR-2332 | QYYZ | NADC30 | SCcd16 | SCya17 | JXA1 1 | VR2332 1 | QYYZ1 | NADC30 1 | |
| Complete genome | 15,319 | 15,451 | 15,526 | 15,047 | 15,321 | 15,315 | 93.1 * | 86.9 * | 85.3 * | 83.6 * |
| 93.2 ** | 87.0 ** | 85.7 ** | 82.0 ** | |||||||
| 5′UTR | 189 | 189 | 190 | 191 | 189 | 188 | 89.4 | 91.4 | 89.4 | 90.7 |
| 87.4 | 88.7 | 85.4 | 85.4 | |||||||
| ORF1a | 7422/2475 | 7512/2502 | 7614/2538 | 7119/2375 | 7422/2475 | 7422/2475 | 96.0/95.3 | 85.3/84.2 | 81.8/82.7 | 76.7/79.1 |
| 94.6/93.1 | 84.8/83.7 | 81.8/83.1 | 76.6/79.0 | |||||||
| ORF1b | 4383/1462 | 4383/1462 | 4383/1462 | 4383/1462 | 4383/1462 | 4383/1462 | 93.4/97.7 | 89.3/95.8 | 88.8/95.9 | 90.1/97.1 |
| 95.3/97.7 | 89.3/95.9 | 89.0/95.6 | 87.7/95.9 | |||||||
| ORF2 | 771/256 | 771/256 | 771/256 | 771/256 | 771/256 | 771/256 | 86.5/85.6 | 87.7/88.7 | 87.4/89.9 | 94.2/93.4 |
| 86.9/86.0 | 89.4/88.7 | 89.4/90.7 | 89.8/92.2 | |||||||
| ORF3 | 765/254 | 765/254 | 765/254 | 765/254 | 765/254 | 765/254 | 80.9/74.9 | 81.8/76.9 | 79.2/75.3 | 92.9/87.5 |
| 85.5/83.5 | 88.0/88.2 | 87.2/85.1 | 83.5/81.6 | |||||||
| ORF4 | 537/178 | 537/178 | 537/178 | 537/178 | 537/178 | 537/178 | 82.7/77.1 | 85.7/78.2 | 83.6/78.2 | 90.5/87.2 |
| 86.8/88.8 | 88.6/90.5 | 87.7/89.9 | 85.3/86.6 | |||||||
| ORF5 | 603/200 | 603/200 | 603/200 | 603/200 | 603/200 | 603/200 | 84.2/82.1 | 84.6/81.1 | 93.4/93.0 | 85.4/83.6 |
| 83.7/82.6 | 85.1/82.6 | 92.0/94.0 | 83.6/84.6 | |||||||
| ORF6 | 525/174 | 525/174 | 525/174 | 525/174 | 525/174 | 525/174 | 91.2/94.3 | 92.2/95.4 | 95.0/97.1 | 89.1/92.0 |
| 91.4/95.4 | 92.0/95.4 | 93.5/98.3 | 89.7/93.1 | |||||||
| ORF7 | 372/123 | 372/123 | 372/123 | 372/123 | 372/123 | 372/123 | 90.9/89.5 | 93.0/92.7 | 93.5/95.2 | 89.2/89.5 |
| 89.8/89.5 | 91.7/91.1 | 92.2/94.4 | 88.7/90.3 | |||||||
| 3′UTR | 150 | 151 | 151 | 151 | 151 | 146 | 99.5 | 92.1 | 94.2 | 92.1 |
| 97.4 | 91.6 | 93.7 | 89.5 | |||||||
| Recombinant Strain | Breakpoints | Parental Sequence | Detection Methods (p-Value) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Beginning | Ending | Minor | Major | RDP | GENECONV | BootScan | MaxChi | Chimaera | SiScan | 3Seq | |
| SCcd16 | 10,401 | 13,841 | NADC30 | JXA1 | 2.120 × 10−51 | 1.358 × 10−13 | 2.281 × 10−52 | 5.308 × 10−21 | 2.234 × 10−23 | 1.473 × 10−32 | 1.069 × 10−120 |
| 13,842 | 15,281 | FJFS | JXA1 | 5.691 × 10−26 | NS | 5.511 × 10−16 | 5.874 × 10−19 | 2.155 × 10−11 | 3.554 × 10−21 | 2.593 × 10−34 | |
| SCya17 | 11,603 | 12,498 | NADC30 | JXA1 | 2.845 × 10−40 | 3.323 × 10−14 | 5.417 × 10−42 | 7.336 × 10−14 | 1.030 × 10−15 | 1.684 × 10−08 | 1.907 × 10−15 |
| 12,690 | 14,777 | XJzx1-2015 | JXA1 | 1.374 × 10−49 | NS | 5.013 × 10−32 | 2.658 × 10−18 | 2.064 × 10−21 | 8.414 × 10−17 | 3.349 × 10−29 | |
| Strains | Isolation Date | Recombination Pattern (Major Parent + Minor Parent) | Virulence | Accession No. |
|---|---|---|---|---|
| GM2 | 2011 | JXA1-P80 (lineage 8) + QYYZ (lineage 3) | Low Pathogenic | JN662424 |
| HENAN-HEB | 2013 | NADC30 (lineage 1) + JXA1 (lineage 8) | - | KJ143621 |
| HENAN-XINX | 2013 | NADC30 (lineage 1) + VR-2332 (lineage 5) | - | KF611905 |
| CHsx1401 | 2014 | NADC30 (lineage 1) + VR-2332 (lineage 5) | Moderately Pathogenic | KP861625 |
| JL580 | 2014 | NADC30 (lineage 1) + 09NEN1 (lineage 8) | Highly Pathogenic | KR706343 |
| FJ1402 | 2014 | NADC30 (lineage 1) + GD (lineage 8) | Highly Pathogenic | KX169191 |
| GD1404 | 2014 | JXA1-P80 (lineage 8) + QYYZ (lineage 3) | Highly Pathogenic | MF124329 |
| GDsg | 2015 | JXA1-P80 (lineage 8) + QYYZ (lineage 3) | Moderately Pathogenic | KX621003 |
| HNyc15 | 2015 | NADC30 (lineage 1) + VR-2332 (lineage 5) | - | KT945018 |
| 15HEN1 | 2015 | NADC30 (lineage 1) + JXA1-P80 (lineage 8) | - | KX815413 |
| SDhz1512 | 2015 | JXA1 (lineage 8) + VR-2332 (lineage 5) + NADC30 (lineage 1) | Low Pathogenic | KX980392 |
| TJnh1501 | 2015 | CHsx1401 (lineage 1) + TJbd14-1 (lineage 8) | Moderately Pathogenic | KX510269 |
| HNhx | 2016 | NADC30 (lineage 1) + JXA1 (lineage 8) | - | KX766379 |
| SCcd17 | 2017 | NADC30 (lineage 1) + JXA1 (lineage 8) + VR-2332 (lineage 5) | - | MG914067 |
| SCcd16 | 2016 | JXA1 (lineage 8) + NADC30 (lineage 1) + QYYZ (lineage 3) | - | MF196905 |
| SCya17 | 2017 | JXA1 (lineage 8) + NADC30 (lineage 1) + QYYZ (lineage 3) | - | MH324400 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Kang, R.; Zhang, Y.; Ding, M.; Xie, B.; Tian, Y.; Wu, X.; Zuo, L.; Yang, X.; Wang, H. Whole Genome Analysis of Two Novel Type 2 Porcine Reproductive and Respiratory Syndrome Viruses with Complex Genome Recombination between Lineage 8, 3, and 1 Strains Identified in Southwestern China. Viruses 2018, 10, 328. https://doi.org/10.3390/v10060328
Zhou L, Kang R, Zhang Y, Ding M, Xie B, Tian Y, Wu X, Zuo L, Yang X, Wang H. Whole Genome Analysis of Two Novel Type 2 Porcine Reproductive and Respiratory Syndrome Viruses with Complex Genome Recombination between Lineage 8, 3, and 1 Strains Identified in Southwestern China. Viruses. 2018; 10(6):328. https://doi.org/10.3390/v10060328
Chicago/Turabian StyleZhou, Long, Runmin Kang, Yi Zhang, Mengdie Ding, Bo Xie, Yiming Tian, Xuan Wu, Lei Zuo, Xin Yang, and Hongning Wang. 2018. "Whole Genome Analysis of Two Novel Type 2 Porcine Reproductive and Respiratory Syndrome Viruses with Complex Genome Recombination between Lineage 8, 3, and 1 Strains Identified in Southwestern China" Viruses 10, no. 6: 328. https://doi.org/10.3390/v10060328
APA StyleZhou, L., Kang, R., Zhang, Y., Ding, M., Xie, B., Tian, Y., Wu, X., Zuo, L., Yang, X., & Wang, H. (2018). Whole Genome Analysis of Two Novel Type 2 Porcine Reproductive and Respiratory Syndrome Viruses with Complex Genome Recombination between Lineage 8, 3, and 1 Strains Identified in Southwestern China. Viruses, 10(6), 328. https://doi.org/10.3390/v10060328