Tick-Borne Flaviviruses and the Type I Interferon Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
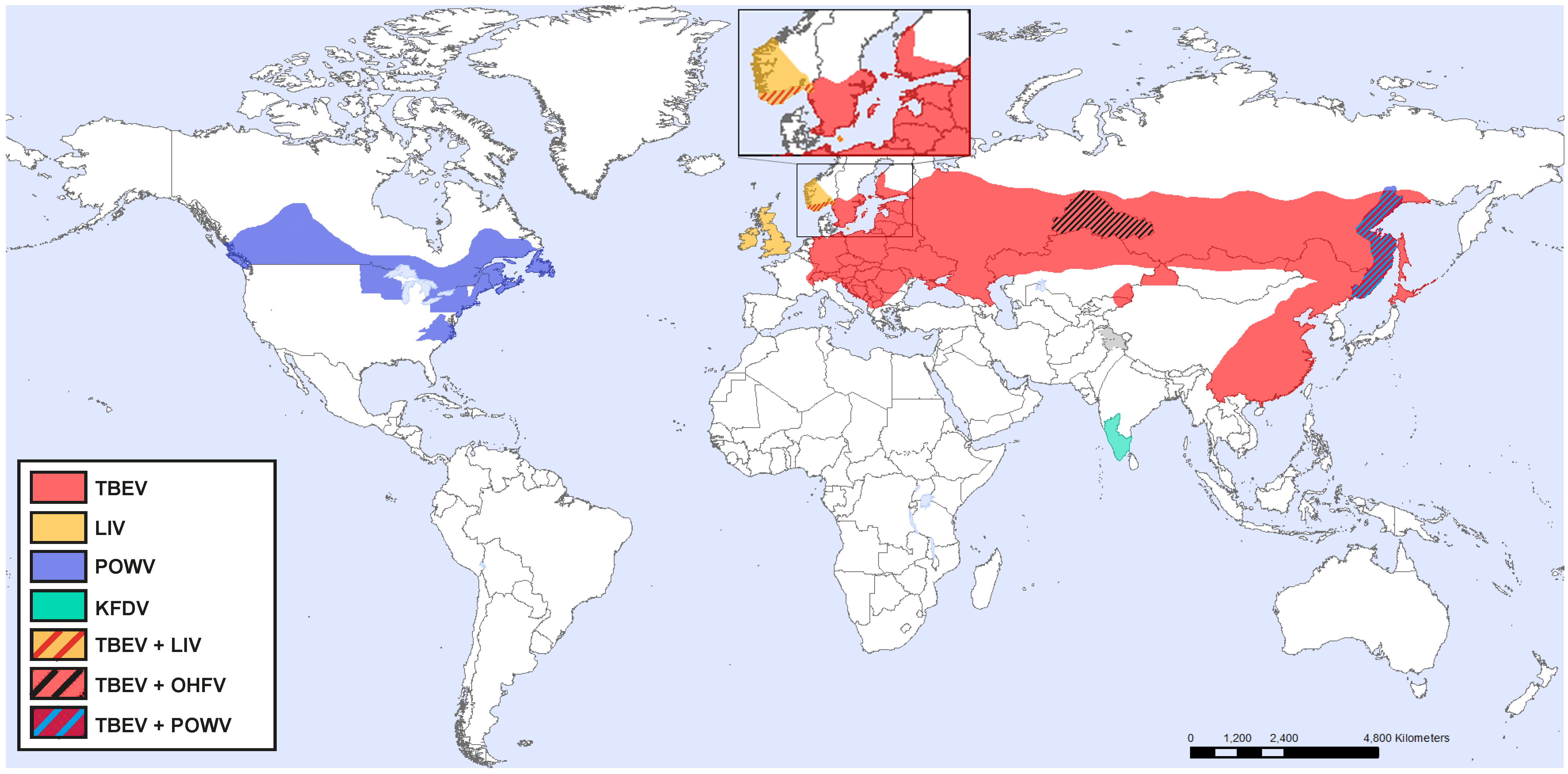
1.1. Tick-Borne Encephalitis Virus
1.2. Powassan Virus
1.3. Louping-Ill Virus
1.4. Omsk Hemorrhagic Fever Virus
1.5. Kyasanur Forest Disease Virus
2. TBFV Genomic Organization
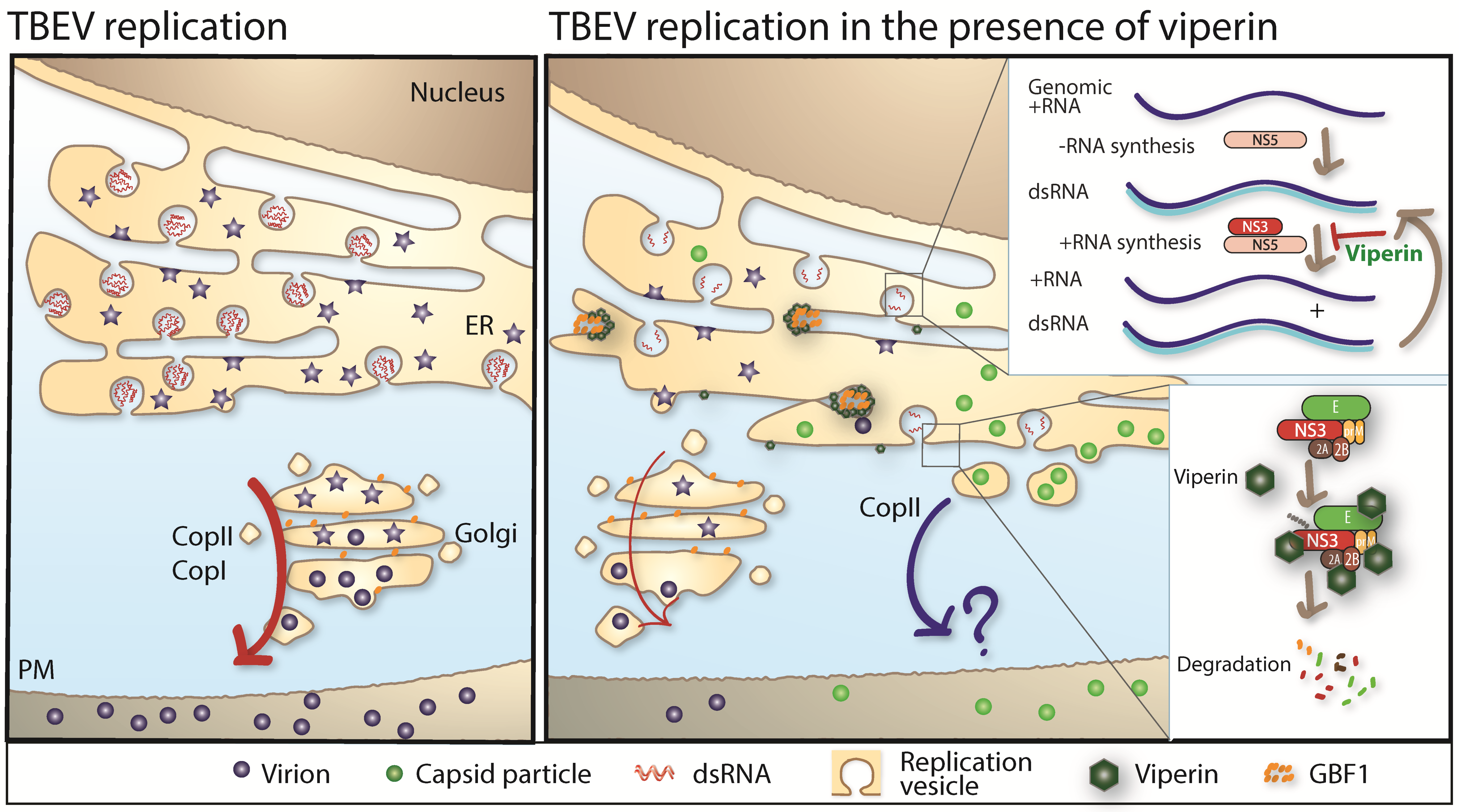
3. Replication Cycle of TBFV
4. Tick–Virus Interactions
5. The Innate Immune Response
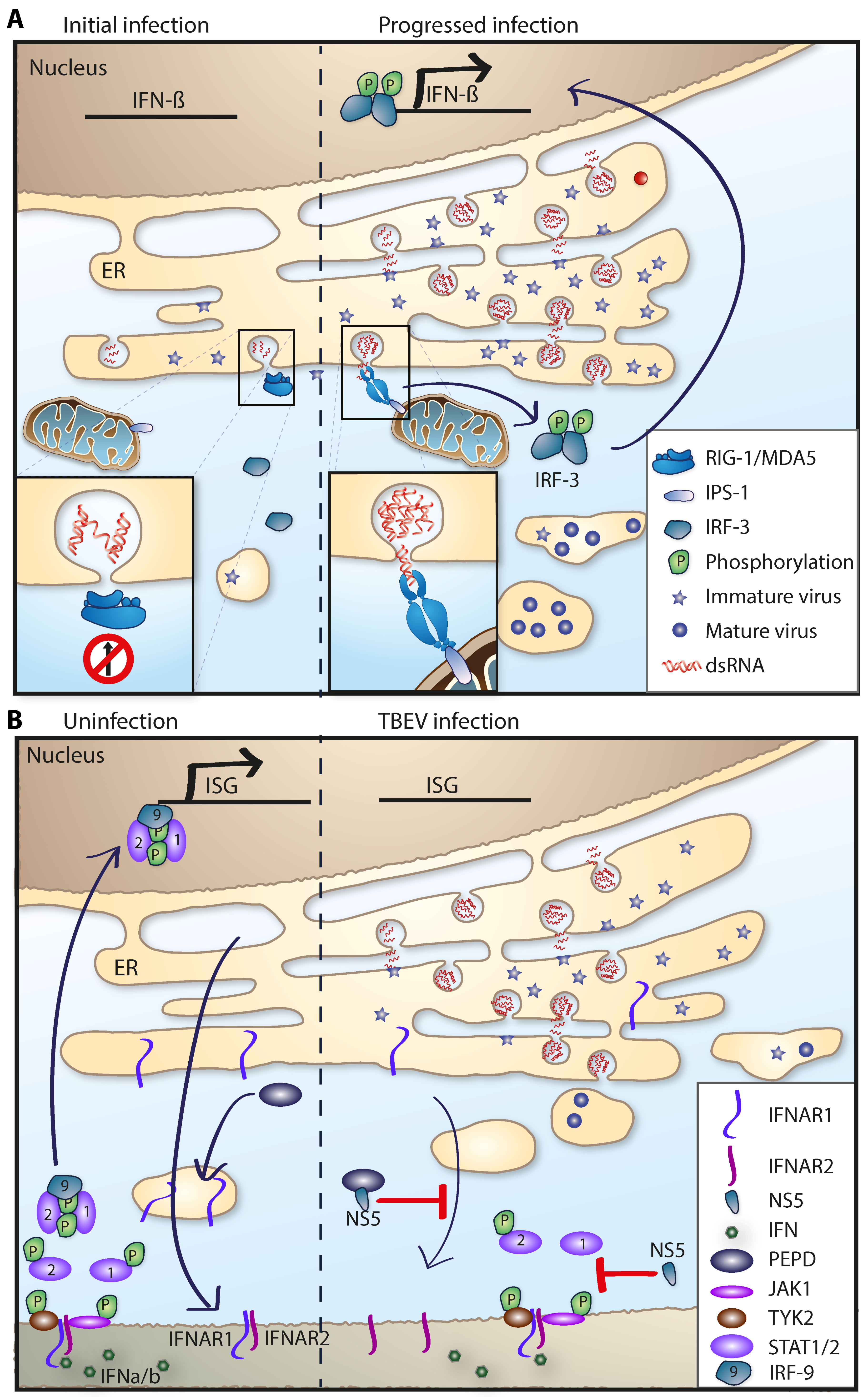
5.1. The Type I IFN Response
5.1.1. The Role of RIG-I-Like and Toll-Like Receptor Signaling
5.1.2. Interferon Signaling and the ISG Response
5.1.3. TBFV: Evasion and Antagonism of the Innate Immune Response
6. Future Considerations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fajardo, A.; Cristina, J.; Moreno, P. Emergence and spreading potential of zika virus. Front. Microbiol. 2016, 7, 1667. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Jamdar, S.F.; Alalowi, M.; Al Ageel Al Beaiji, S.M. Dengue virus: A global human threat: Review of literature. J. Int. Soc. Prev. Community Dent. 2016, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Martin-Acebes, M.A.; Saiz, J.C. West nile virus: A re-emerging pathogen revisited. World J. Virol. 2012, 1, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, A.L.; McClain, D. Alphaviruses (togaviridae) and flaviviruses (flaviviridae). In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Kim, C.W.; Chang, K.M. Hepatitis C virus: Virology and life cycle. Clin. Mol. Hepatol. 2013, 19, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Babenko, L.V.; Davydova, M.S.; Zakorkina, T.N.; Blokhin; Voronkov, N.A.; Naumov, R.L.; Khizhinskii, P.G. Characteristics of a nidus of tick-borne encephalitis in the construction zone of the krasnoyarsk hydroelectric station and development of measures for protection of workers against ticks; preliminary report. Med. Parazitol. 1958, 27, 6–14. [Google Scholar]
- Brackney, D.E.; Nofchissey, R.A.; Fitzpatrick, K.A.; Brown, I.K.; Ebel, G.D. Stable prevalence of powassan virus in ixodes scapularis in a northern wisconsin focus. Am. J. Trop. Med. Hyg. 2008, 79, 971–973. [Google Scholar] [PubMed]
- Kramer, L.D.; Styer, L.M.; Ebel, G.D. A global perspective on the epidemiology of west nile virus. Annu. Rev. Entomol. 2008, 53, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. Zika virus: New clinical syndromes and its emergence in the western hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.O.; Mertens, E.; Despres, P. West nile virus and its emergence in the united states of america. Vet. Res. 2010, 41, 67. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T. Flavivirus encephalitis. N. Engl. J. Med. 2004, 351, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J. The changing epidemiology of yellow fever and dengue, 1900 to 2003: Full circle? Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.D.; Higgs, S. Yellow fever: A disease that has yet to be conquered. Annu. Rev. Entomol. 2007, 52, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.J.; Oduyebo, T.; Brault, A.C.; Brooks, J.T.; Chung, K.W.; Hills, S.; Kuehnert, M.J.; Mead, P.; Meaney-Delman, D.; Rabe, I.; et al. Modes of transmission of zika virus. J. Infect. Dis. 2017, 216, S875–S883. [Google Scholar] [CrossRef] [PubMed]
- Parola, P.; Raoult, D. Ticks and tickborne bacterial diseases in humans: An emerging infectious threat. Clin. Infect. Dis. 2001, 32, 897–928. [Google Scholar] [CrossRef] [PubMed]
- Ebel, G.D.; Kramer, L.D. Short report: Duration of tick attachment required for transmission of powassan virus by deer ticks. Am. J. Trop. Med. Hyg. 2004, 71, 268–271. [Google Scholar] [PubMed]
- Dobler, G. Zoonotic tick-borne flaviviruses. Vet. Microbiol. 2010, 140, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, P.A. Molecular characterization of tick-virus interactions. Front. Biosci. 2009, 14, 2466–2483. [Google Scholar] [CrossRef]
- Nuttall, P.A.; Labuda, M. Dynamics of infection in tick vectors and at the tick-host interface. Adv. Virus Res. 2003, 60, 233–272. [Google Scholar] [PubMed]
- Labuda, M.; Nuttall, P.A.; Kozuch, O.; Eleckova, E.; Williams, T.; Zuffova, E.; Sabo, A. Non-viraemic transmission of tick-borne encephalitis virus: A mechanism for arbovirus survival in nature. Experientia 1993, 49, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Ecker, M.; Allison, S.L.; Meixner, T.; Heinz, F.X. Sequence analysis and genetic classification of tick-borne encephalitis viruses from europe and asia. J. Gen. Virol. 1999, 80, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Gritsun, T.S.; Nuttall, P.A.; Gould, E.A. Tick-borne flaviviruses. Adv. Virus Res. 2003, 61, 317–371. [Google Scholar] [PubMed]
- Lindquist, L.; Vapalahti, O. Tick-borne encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef]
- Dobler, G.; Gniel, D.; Petermann, R.; Pfeffer, M. Epidemiology and distribution of tick-borne encephalitis. Wien. Med. Wochenschr. 2012, 162, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Suss, J. Tick-borne encephalitis 2010: Epidemiology, risk areas, and virus strains in europe and asia-an overview. Ticks Tick Borne Dis. 2011, 2, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Soleng, A.; Edgar, K.S.; Paulsen, K.M.; Pedersen, B.N.; Okbaldet, Y.B.; Skjetne, I.E.B.; Gurung, D.; Vikse, R.; Andreassen, A.K. Distribution of ixodes ricinus ticks and prevalence of tick-borne encephalitis virus among questing ticks in the arctic circle region of northern norway. Ticks Tick Borne Dis. 2018, 9, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, I.; Bassetti, M.; Paladini, G.; Luzzati, R.; Santon, D.; Merelli, M.; Sabbata, G.D.; Carletti, T.; Marcello, A.; D’Agaro, P. Persistent viremia and urine shedding of tick-borne encephalitis virus in an infected immunosuppressed patient from a new epidemic cluster in north-eastern italy. J. Clin. Virol. 2015, 69, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Jaenson, T.G.; Hjertqvist, M.; Bergstrom, T.; Lundkvist, A. Why is tick-borne encephalitis increasing? A review of the key factors causing the increasing incidence of human tbe in sweden. Parasit. Vectors 2012, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Velay, A.; Solis, M.; Kack-Kack, W.; Gantner, P.; Maquart, M.; Martinot, M.; Augereau, O.; De Briel, D.; Kieffer, P.; Lohmann, C.; et al. A new hot spot for tick-borne encephalitis (tbe): A marked increase of tbe cases in france in 2016. Ticks Tick Borne Dis. 2018, 9, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Randolph, S.E. To what extent has climate change contributed to the recent epidemiology of tick-borne diseases? Vet. Parasitol. 2010, 167, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Sumilo, D.; Bormane, A.; Asokliene, L.; Vasilenko, V.; Golovljova, I.; Avsic-Zupanc, T.; Hubalek, Z.; Randolph, S.E. Socio-economic factors in the differential upsurge of tick-borne encephalitis in central and eastern europe. Rev. Med. Virol. 2008, 18, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Labuda, M.; Randolph, S.E. Survival strategy of tick-borne encephalitis virus: Cellular basis and environmental determinants. Zentralbl. Bakteriol. 1999, 289, 513–524. [Google Scholar] [CrossRef]
- Mansfield, K.L.; Johnson, N.; Phipps, L.P.; Stephenson, J.R.; Fooks, A.R.; Solomon, T. Tick-borne encephalitis virus—A review of an emerging zoonosis. J. Gen. Virol. 2009, 90, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Markovinovic, L.; Kosanovic Licina, M.L.; Tesic, V.; Vojvodic, D.; Vladusic Lucic, I.; Kniewald, T.; Vukas, T.; Kutlesa, M.; Krajinovic, L.C. An outbreak of tick-borne encephalitis associated with raw goat milk and cheese consumption, croatia, 2015. Infection 2016, 44, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Hudopisk, N.; Korva, M.; Janet, E.; Simetinger, M.; Grgic-Vitek, M.; Gubensek, J.; Natek, V.; Kraigher, A.; Strle, F.; Avsic-Zupanc, T. Tick-borne encephalitis associated with consumption of raw goat milk, slovenia, 2012. Emergy Infect. Dis. 2013, 19, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Lipowski, D.; Popiel, M.; Perlejewski, K.; Nakamura, S.; Bukowska-Osko, I.; Rzadkiewicz, E.; Dzieciatkowski, T.; Milecka, A.; Wenski, W.; Ciszek, M.; et al. A cluster of fatal tick-borne encephalitis virus infection in organ transplant setting. J. Infect. Dis. 2017, 215, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.J.; Mitzel, D.N.; Taylor, R.T.; Best, S.M.; Bloom, M.E. Tick-borne flaviviruses: Dissecting host immune responses and virus countermeasures. Immunol. Res. 2009, 43, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.J.; Halliday, G.M.; King, N.J. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J. Investig. Dermatol. 2000, 114, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, S.; Schlesinger, M.J. The Togaviridae and Flaviviridae; Plenum Press: New York, NY, USA, 1986; p. 453. [Google Scholar]
- Ruzek, D.; Dobler, G.; Donoso Mantke, O. Tick-borne encephalitis: Pathogenesis and clinical implications. Travel Med. Infect. Dis. 2010, 8, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R. The clinical and epidemiological profile of tick-borne encephalitis in southern germany 1994–1998: A prospective study of 656 patients. Brain 1999, 122, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Mickiene, A.; Laiskonis, A.; Gunther, G.; Vene, S.; Lundkvist, A.; Lindquist, L. Tickborne encephalitis in an area of high endemicity in lithuania: Disease severity and long-term prognosis. Clin. Infect. Dis. 2002, 35, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Henningsson, A.J.; Lindqvist, R.; Norberg, P.; Lindblom, P.; Roth, A.; Forsberg, P.; Bergstrom, T.; Overby, A.K.; Lindgren, P.E. Human tick-borne encephalitis and characterization of virus from biting tick. Emerg. Infect. Dis. 2016, 22, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Dorrbecker, B.; Dobler, G.; Spiegel, M.; Hufert, F.T. Tick-borne encephalitis virus and the immune response of the mammalian host. Travel Med. Infect. Dis. 2010, 8, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Zambito Marsala, S.; Pistacchi, M.; Gioulis, M.; Mel, R.; Marchini, C.; Francavilla, E. Neurological complications of tick borne encephalitis: The experience of 89 patients studied and literature review. Neurol. Sci. 2014, 35, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Haglund, M.; Gunther, G. Tick-borne encephalitis—Pathogenesis, clinical course and long-term follow-up. Vaccine 2003, 21 (Suppl. 1), S11–S18. [Google Scholar] [CrossRef]
- Lani, R.; Moghaddam, E.; Haghani, A.; Chang, L.Y.; AbuBakar, S.; Zandi, K. Tick-borne viruses: A review from the perspective of therapeutic approaches. Ticks Tick Borne Dis. 2014, 5, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.A.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Woodall, J.P.; Roz, A. Experimental milk-borne transmission of powassan virus in the goat. Am. J. Trop. Med. Hyg. 1977, 26, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Hermance, M.E.; Thangamani, S. Tick saliva enhances powassan virus transmission to the host, influencing its dissemination and the course of disease. J. Virol. 2015, 89, 7852–7860. [Google Scholar] [CrossRef] [PubMed]
- Hermance, M.E.; Santos, R.I.; Kelly, B.C.; Valbuena, G.; Thangamani, S. Immune cell targets of infection at the tick-skin interface during powassan virus transmission. PLoS ONE 2016, 11, e0155889. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.I.; Hermance, M.E.; Gelman, B.B.; Thangamani, S. Spinal cord ventral horns and lymphoid organ involvement in powassan virus infection in a mouse model. Viruses 2016, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Gholam, B.I.; Puksa, S.; Provias, J.P. Powassan encephalitis: A case report with neuropathology and literature review. CMAJ 1999, 161, 1419–1422. [Google Scholar] [PubMed]
- Ebel, G.D. Update on powassan virus: Emergence of a north american tick-borne flavivirus. Annu. Rev. Entomol. 2010, 55, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Hermance, M.E.; Thangamani, S. Powassan virus: An emerging arbovirus of public health concern in north america. Vector Borne Zoonotic Dis. 2017, 17, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L. Louping ill virus in the UK: A review of the hosts, transmission and ecological consequences of control. Exp. Appl. Acarol. 2016, 68, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, C.L.; Mansfield, K.L.; Phipps, L.P.; Wakeley, P.R.; Mearns, R.; Schock, A.; Bell, S.; Breed, A.C.; Fooks, A.R.; Johnson, N. Louping ill virus: An endemic tick-borne disease of great britain. J. Gen. Virol. 2014, 95, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Rivers, T.M.; Schwentker, F.F. Louping ill in man. J. Exp. Med. 1934, 59, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.H.; Manderson, W.G.; Hurst, E.W. Louping-ill meningo-encephalitis; a further case and a serological survey. Lancet 1949, 2, 696–699. [Google Scholar] [CrossRef]
- Schonell, M.E.; Brotherston, J.G.; Burnett, R.C.; Campbell, J.; Coghlan, J.D.; Moffat, M.A.; Norval, J.; Sutherland, J.A. Occupational infections in the edinburgh abattoir. Br. Med. J. 1966, 2, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Reid, H.W.; Gibbs, C.A.; Burrells, C.; Doherty, P.C. Laboratory infections with louping-ill virus. Lancet 1972, 1, 592–593. [Google Scholar] [CrossRef]
- Davidson, M.M.; Williams, H.; Macleod, J.A. Louping ill in man: A forgotten disease. J. Infect. 1991, 23, 241–249. [Google Scholar] [CrossRef]
- Ruzek, D.; Yakimenko, V.V.; Karan, L.S.; Tkachev, S.E. Omsk haemorrhagic fever. Lancet 2010, 376, 2104–2113. [Google Scholar] [CrossRef]
- Trapido, H.; Rajagopalan, P.K.; Work, T.H.; Varma, M.G. Kyasanur forest disease. VIII. Isolation of kyasanur forest disease virus from naturally infected ticks of the genus haemaphysalis. Indian J. Med. Res. 1959, 47, 133–138. [Google Scholar] [PubMed]
- Holbrook, M.R. Kyasanur forest disease. Antivir. Res. 2012, 96, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavri, K. Clinical, clinicopathologic, and hematologic features of kyasanur forest disease. Rev. Infect. Dis. 1989, 11 (Suppl. 4), S854–S859. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, P. Kyasanur forest disease: An epidemiological view in India. Rev. Med. Virol. 2006, 16, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Dandawate, C.N.; Desai, G.B.; Achar, T.R.; Banerjee, K. Field evaluation of formalin inactivated kyasanur forest disease virus tissue culture vaccine in three districts of karnataka state. Indian J. Med. Res. 1994, 99, 152–158. [Google Scholar] [PubMed]
- Kasabi, G.S.; Murhekar, M.V.; Sandhya, V.K.; Raghunandan, R.; Kiran, S.K.; Channabasappa, G.H.; Mehendale, S.M. Coverage and effectiveness of kyasanur forest disease (KFD) vaccine in karnataka, south india, 2005–2010. PLoS Negl. Trop. Dis. 2013, 7, e2025. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Blanco, M.A.; Vasudevan, S.G.; Bradrick, S.S.; Nicchitta, C. Flavivirus RNA transactions from viral entry to genome replication. Antivir. Res. 2016, 134, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Blitvich, B.J.; Firth, A.E. Insect-specific flaviviruses: A systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses 2015, 7, 1927–1959. [Google Scholar] [CrossRef] [PubMed]
- Kroschewski, H.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Role of heparan sulfate for attachment and entry of tick-borne encephalitis virus. Virology 2003, 308, 92–100. [Google Scholar] [CrossRef]
- Kaufmann, B.; Rossmann, M.G. Molecular mechanisms involved in the early steps of flavivirus cell entry. Microbes Infect. 2011, 13, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013. [Google Scholar]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Klema, V.J.; Padmanabhan, R.; Choi, K.H. Flaviviral replication complex: Coordination between RNA synthesis and 5′-RNA capping. Viruses 2015, 7, 4640–4656. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, S.; Reid, D.W.; Cox, A.H.; Nicchitta, C.V. De novo translation initiation on membrane-bound ribosomes as a mechanism for localization of cytosolic protein mrnas to the endoplasmic reticulum. RNA 2014, 20, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS ONE 2012, 7, e47912. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Popov, V.L.; Niedrig, M.; Weber, F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 2010, 84, 8470–8483. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Weber, F. Hiding from intracellular pattern recognition receptors, a passive strategy of flavivirus immune evasion. Virulence 2011, 2, 238–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Albornoz, A.; Baba, M.M.; D’Agaro, P.; Marcello, A. Formation of membrane-defined compartments by tick-borne encephalitis virus contributes to the early delay in interferon signaling. Virus Res. 2012, 163, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Maiuri, P.; Hoenninger, V.M.; Mandl, C.W.; Marcello, A. Spatial and temporal organization of tick-borne encephalitis flavivirus replicated RNA in living cells. Virology 2008, 379, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Maiuri, P.; Marcello, A. Visual detection of flavivirus RNA in living cells. Methods 2016, 98, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Vonderstein, K.; Nilsson, E.; Hubel, P.; Nygard Skalman, L.; Upadhyay, A.; Pasto, J.; Pichlmair, A.; Lundmark, R.; Overby, A.K. Viperin targets flavivirus virulence by inducing assembly of non-infectious capsid particles. J. Virol. 2017. [Google Scholar] [CrossRef]
- Yoshii, K.; Yanagihara, N.; Ishizuka, M.; Sakai, M.; Kariwa, H. N-linked glycan in tick-borne encephalitis virus envelope protein affects viral secretion in mammalian cells, but not in tick cells. J. Gen. Virol. 2013, 94, 2249–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of dengue virus: Implications for flavivirus organization, maturation, and fusion. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef]
- Yu, I.M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the immature dengue virus at low ph primes proteolytic maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Elshuber, S.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Cleavage of protein prm is necessary for infection of bhk-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 2003, 84, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar] [PubMed]
- Mackenzie, J.M.; Westaway, E.G. Assembly and maturation of the flavivirus kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef] [PubMed]
- Schnettler, E.; Tykalova, H.; Watson, M.; Sharma, M.; Sterken, M.G.; Obbard, D.J.; Lewis, S.H.; McFarlane, M.; Bell-Sakyi, L.; Barry, G.; et al. Induction and suppression of tick cell antiviral RNAi responses by tick-borne flaviviruses. Nucleic Acids Res. 2014, 42, 9436–9446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisheit, S.; Villar, M.; Tykalova, H.; Popara, M.; Loecherbach, J.; Watson, M.; Ruzek, D.; Grubhoffer, L.; de la Fuente, J.; Fazakerley, J.K.; et al. Ixodes scapularis and ixodes ricinus tick cell lines respond to infection with tick-borne encephalitis virus: Transcriptomic and proteomic analysis. Parasit Vectors 2015, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Basset, C.; Holton, J.; O’Mahony, R.; Roitt, I. Innate immunity and pathogen-host interaction. Vaccine 2003, 21 (Suppl. 2), S12–S23. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Finsterbusch, K.; Lindquist, R.; Nair, S.; Lienenklaus, S.; Gekara, N.O.; Janik, D.; Weiss, S.; Kalinke, U.; Overby, A.K.; et al. Type I interferon protects mice from fatal neurotropic infection with langat virus by systemic and local antiviral responses. J. Virol. 2014, 88, 12202–12212. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, R.; Mundt, F.; Gilthorpe, J.D.; Wolfel, S.; Gekara, N.O.; Kroger, A.; Overby, A.K. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflamm. 2016, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Stancek, D.; Vilcek, J. The role of interferon in tick-borne encephalitis virus-infected l cells. I. Acute infection. Acta Virol. 1965, 9, 1–8. [Google Scholar] [PubMed]
- Vilcek, J. An interferon-like substance released from tickborne encephalitis virus-infected chick embryo fibroblast cells. Nature 1960, 187, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Flint, M.; McMullan, L.K.; Dodd, K.A.; Bird, B.H.; Khristova, M.L.; Nichol, S.T.; Spiropoulou, C.F. Inhibitors of the tick-borne, hemorrhagic fever-associated flaviviruses. Antimicrob. Agents Chemother. 2014, 58, 3206–3216. [Google Scholar] [CrossRef] [PubMed]
- Mlera, L.; Meade-White, K.; Dahlstrom, E.; Baur, R.; Kanakabandi, K.; Virtaneva, K.; Porcella, S.F.; Bloom, M.E. Peromyscus leucopus mouse brain transcriptome response to powassan virus infection. J. Neurovirol. 2018, 24, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.L.; Johnson, N.; Banyard, A.C.; Nunez, A.; Baylis, M.; Solomon, T.; Fooks, A.R. Innate and adaptive immune responses to tick-borne flavivirus infection in sheep. Vet. Microbiol. 2016, 185, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.A.; Bird, B.H.; Jones, M.E.; Nichol, S.T.; Spiropoulou, C.F. Kyasanur forest disease virus infection in mice is associated with higher morbidity and mortality than infection with the closely related alkhurma hemorrhagic fever virus. PLoS ONE 2014, 9, e100301. [Google Scholar] [CrossRef] [PubMed]
- De Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Recognition of viruses by innate immunity. Immunol. Rev. 2007, 220, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Van Boxel-Dezaire, A.H.; Rani, M.R.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- De Veer, M.J.; Holko, M.; Frevel, M.; Walker, E.; Der, S.; Paranjape, J.M.; Silverman, R.H.; Williams, B.R. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 2001, 69, 912–920. [Google Scholar] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of nf-kappab by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Liu, P.; Latz, E.; Golenbock, D.T.; Finberg, R.W.; Libraty, D.H. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of tlr7 signaling beyond endosomal recognition. J. Immunol. 2006, 177, 7114–7121. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase rig-i has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-triphosphate RNA is the ligand for rig-i. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Gale, M., Jr. Differential recognition of double-stranded RNA by rig-i-like receptors in antiviral immunity. J. Exp. Med. 2008, 205, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor trif in the myd88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through toll-like receptors involves a direct interaction of irf7 with myd88 and traf6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of mavs, a mitochondrial antiviral signaling protein that activates nf-kappab and irf 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. Ips-1, an adaptor triggering rig-i- and mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. Visa is an adapter protein required for virus-triggered ifn-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the rig-i antiviral pathway and is targeted by hepatitis c virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. Irfs: Master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Kurhade, C.; Zegenhagen, L.; Weber, E.; Nair, S.; Michaelsen-Preusse, K.; Spanier, J.; Gekara, N.O.; Kroger, A.; Overby, A.K. Type I interferon response in olfactory bulb, the site of tick-borne flavivirus accumulation, is primarily regulated by ips-1. J. Neuroinflamm. 2016, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Carletti, T.; Zakaria, M.K.; Marcello, A. The host cell response to tick-borne encephalitis virus. Biochem. Biophys. Res. Commun. 2017, 492, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, A.; Carletti, T.; Corazza, G.; Marcello, A. The stress granule component tia-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation. J. Virol. 2014, 88, 6611–6622. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.G.; Woods, T.A.; Butchi, N.B.; Morgan, T.M.; Taylor, R.T.; Sunyakumthorn, P.; Mukherjee, P.; Lubick, K.J.; Best, S.M.; Peterson, K.E. Toll-like receptor 7 suppresses virus replication in neurons but does not affect viral pathogenesis in a mouse model of langat virus infection. J. Gen. Virol. 2013, 94, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Barkhash, A.V.; Voevoda, M.I.; Romaschenko, A.G. Association of single nucleotide polymorphism rs3775291 in the coding region of the tlr3 gene with predisposition to tick-borne encephalitis in a russian population. Antivir. Res. 2013, 99, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Grygorczuk, S.; Parczewski, M.; Swierzbinska, R.; Czupryna, P.; Moniuszko, A.; Dunaj, J.; Kondrusik, M.; Pancewicz, S. The increased concentration of macrophage migration inhibitory factor in serum and cerebrospinal fluid of patients with tick-borne encephalitis. J. Neuroinflamm. 2017, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Kindberg, E.; Vene, S.; Mickiene, A.; Lundkvist, A.; Lindquist, L.; Svensson, L. A functional toll-like receptor 3 gene (tlr3) may be a risk factor for tick-borne encephalitis virus (tbev) infection. J. Infect. Dis. 2011, 203, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Mickiene, A.; Pakalniene, J.; Nordgren, J.; Carlsson, B.; Hagbom, M.; Svensson, L.; Lindquist, L. Polymorphisms in chemokine receptor 5 and toll-like receptor 3 genes are risk factors for clinical tick-borne encephalitis in the lithuanian population. PLoS ONE 2014, 9, e106798. [Google Scholar] [CrossRef] [PubMed]
- Ranjith-Kumar, C.T.; Miller, W.; Sun, J.; Xiong, J.; Santos, J.; Yarbrough, I.; Lamb, R.J.; Mills, J.; Duffy, K.E.; Hoose, S.; et al. Effects of single nucleotide polymorphisms on toll-like receptor 3 activity and expression in cultured cells. J. Biol. Chem. 2007, 282, 17696–17705. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-i- and type-ii-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, R.; Kurhade, C.; Gilthorpe, J.D.; Overby, A.K. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J. Neuroinflamm. 2018, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Preusser, M.; Garzuly, F.; Holzmann, H.; Heinz, F.X.; Budka, H. Visualization of central european tick-borne encephalitis infection in fatal human cases. J. Neuropathol. Exp. Neurol. 2005, 64, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Kornyey, S. Contribution to the histology of tick-borne encephalitis. Acta Neuropathol. 1978, 43, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Palus, M.; Bily, T.; Elsterova, J.; Langhansova, H.; Salat, J.; Vancova, M.; Ruzek, D. Infection and injury of human astrocytes by tick-borne encephalitis virus. J. Gen. Virol. 2014, 95, 2411–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potokar, M.; Korva, M.; Jorgacevski, J.; Avsic-Zupanc, T.; Zorec, R. Tick-borne encephalitis virus infects rat astrocytes but does not affect their viability. PLoS ONE 2014, 9, e86219. [Google Scholar] [CrossRef] [PubMed]
- Selinger, M.; Wilkie, G.S.; Tong, L.; Gu, Q.; Schnettler, E.; Grubhoffer, L.; Kohl, A. Analysis of tick-borne encephalitis virus-induced host responses in human cells of neuronal origin and interferon-mediated protection. J. Gen. Virol. 2017, 98, 2043–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.T.; Lubick, K.J.; Robertson, S.J.; Broughton, J.P.; Bloom, M.E.; Bresnahan, W.A.; Best, S.M. Trim79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 2011, 10, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of interferon-stimulated jak-stat signaling by a tick-borne flavivirus and identification of ns5 as an interferon antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.S.; Vonderstein, K.; Pichlmair, A.; Stehling, O.; Bennett, K.L.; Dobler, G.; Guo, J.T.; Superti-Furga, G.; Lill, R.; Overby, A.K.; et al. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical sam domain activity. Cell Microbiol. 2014, 16, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, C.; Lindqvist, R.; Kurhade, C.; Vonderstein, K.; Pasto, J.; Edlund, K.; Upadhyay, A.S.; Overby, A.K. Viperin restricts zika virus and tick-borne encephalitis virus replication by targeting ns3 for proteasomal degradation. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rebouillat, D.; Hovanessian, A.G. The human 2′,5′-oligoadenylate synthetase family: Interferon-induced proteins with unique enzymatic properties. J. Interferon Cytokine Res. 1999, 19, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.N.; Ghosh, A.; Wang, H.W.; Sung, S.S.; Sen, G.C. The nature of the catalytic domain of 2′-5′-oligoadenylate synthetases. J. Biol. Chem. 1999, 274, 25535–25542. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Barkhash, A.V.; Perelygin, A.A.; Babenko, V.N.; Myasnikova, N.G.; Pilipenko, P.I.; Romaschenko, A.G.; Voevoda, M.I.; Brinton, M.A. Variability in the 2′-5′-oligoadenylate synthetase gene cluster is associated with human predisposition to tick-borne encephalitis virus-induced disease. J. Infect. Dis. 2010, 202, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Moritoh, K.; Nagata, N.; Yokozawa, K.; Sakai, M.; Sasaki, N.; Kariwa, H.; Agui, T.; Takashima, I. Susceptibility to flavivirus-specific antiviral response of oas1b affects the neurovirulence of the far-eastern subtype of tick-borne encephalitis virus. Arch. Virol. 2013, 158, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Stoye, J.P.; Saib, A. Trim family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.C.; Cresswell, P. Viperin (cig5), an ifn-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2001, 98, 15125–15130. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Beard, M.R. The role of viperin in the innate antiviral response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M., Jr.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The interferon-inducible gene viperin restricts west nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Carr, J.M.; Calvert, J.K.; Wati, S.; Clarke, J.N.; Eyre, N.S.; Narayana, S.K.; Fiches, G.N.; McCartney, E.M.; Beard, M.R. Viperin is induced following dengue virus type-2 (denv-2) infection and has anti-viral actions requiring the c-terminal end of viperin. PLoS Negl. Trop. Dis. 2013, 7, e2178. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoek, K.H.; Eyre, N.S.; Shue, B.; Khantisitthiporn, O.; Glab-Ampi, K.; Carr, J.M.; Gartner, M.J.; Jolly, L.A.; Thomas, P.Q.; Adikusuma, F.; et al. Viperin is an important host restriction factor in control of zika virus infection. Sci. Rep. 2017, 7, 4475. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Eyre, N.S.; Yip, E.; Narayana, S.; Li, K.; Fiches, G.; McCartney, E.M.; Jangra, R.K.; Lemon, S.M.; Beard, M.R. The antiviral protein viperin inhibits hepatitis c virus replication via interaction with nonstructural protein 5a. Hepatology 2011, 54, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Yaneva, R.; Cresswell, P. Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 2011, 10, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A. The interferon inducible gene: Viperin. J. Interferon Cytokine Res. 2011, 31, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Shaveta, G.; Shi, J.; Chow, V.T.; Song, J. Structural characterization reveals that viperin is a radical s-adenosyl-l-methionine (sam) enzyme. Biochem. Biophys. Res. Commun. 2010, 391, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.S.; Stehling, O.; Panayiotou, C.; Rosser, R.; Lill, R.; Overby, A.K. Cellular requirements for iron-sulfur cluster insertion into the antiviral radical sam protein viperin. J. Biol. Chem. 2017, 292, 13879–13889. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, M.K.; Li, Y.; Cresswell, P.; Modis, Y.; Ealick, S.E. Structural studies of viperin, an antiviral radical sam enzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 6806–6811. [Google Scholar] [CrossRef] [PubMed]
- Honarmand Ebrahimi, K. A unifying view of the broad-spectrum antiviral activity of rsad2 (viperin) based on its radical-sam chemistry. Metallomics 2018, 10, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Bridwell-Rabb, J.; Zhong, A.; Sun, H.G.; Drennan, C.L.; Liu, H.W. A b12-dependent radical sam enzyme involved in oxetanocin a biosynthesis. Nature 2017, 544, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Carpp, L.N.; Rogers, R.S.; Moritz, R.L.; Aitchison, J.D. Quantitative proteomic analysis of host-virus interactions reveals a role for golgi brefeldin a resistance factor 1 (gbf1) in dengue infection. Mol. Cell. Proteom. 2014, 13, 2836–2854. [Google Scholar] [CrossRef] [PubMed]
- Farhat, R.; Seron, K.; Ferlin, J.; Feneant, L.; Belouzard, S.; Goueslain, L.; Jackson, C.L.; Dubuisson, J.; Rouille, Y. Identification of class ii adp-ribosylation factors as cellular factors required for hepatitis c virus replication. Cell. Microbiol. 2016, 18, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, A.H.; Wannee, K.F.; Scholte, F.E.; Goeman, J.J.; Ten Dijke, P.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. A kinome-wide small interfering RNA screen identifies proviral and antiviral host factors in severe acute respiratory syndrome coronavirus replication, including double-stranded RNA-activated protein kinase and early secretory pathway proteins. J. Virol. 2015, 89, 8318–8333. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Neumann, G.; Kawaoka, Y. Role of the gtpase rab1b in ebolavirus particle formation. J. Virol. 2010, 84, 4816–4820. [Google Scholar] [CrossRef] [PubMed]
- Labuda, M.; Jones, L.D.; Williams, T.; Nuttall, P.A. Enhancement of tick-borne encephalitis virus transmission by tick salivary gland extracts. Med. Vet. Entomol. 1993, 7, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Kazimirova, M.; Stibraniova, I. Tick salivary compounds: Their role in modulation of host defences and pathogen transmission. Front. Cell. Infect. Microbiol. 2013, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, Z.; Wang, M.; Cheng, A. Innate immune evasion mediated by flaviviridae non-structural proteins. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Werme, K.; Wigerius, M.; Johansson, M. Tick-borne encephalitis virus ns5 associates with membrane protein scribble and impairs interferon-stimulated jak-stat signalling. Cell. Microbiol. 2008, 10, 696–712. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M. The many faces of the flavivirus NS5 protein in antagonism of type I interferon signaling. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.W.; Cutts, T.A.; Court, D.A.; Theriault, S. The generation of a reverse genetics system for kyasanur forest disease virus and the ability to antagonize the induction of the antiviral state in vitro. Virus Res. 2012, 163, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Park, G.S.; Morris, K.L.; Hallett, R.G.; Bloom, M.E.; Best, S.M. Identification of residues critical for the interferon antagonist function of langat virus ns5 reveals a role for the RNA-dependent RNA polymerase domain. J. Virol. 2007, 81, 6936–6946. [Google Scholar] [CrossRef] [PubMed]
- Lubick, K.J.; Robertson, S.J.; McNally, K.L.; Freedman, B.A.; Rasmussen, A.L.; Taylor, R.T.; Walts, A.D.; Tsuruda, S.; Sakai, M.; Ishizuka, M.; et al. Flavivirus antagonism of type I interferon signaling reveals prolidase as a regulator of ifnar1 surface expression. Cell Host Microbe 2015, 18, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.W.; Ranadheera, C.; Nikiforuk, A.M.; Cutts, T.A.; Kobasa, D.; Court, D.A.; Theriault, S.S. Limited effects of type I interferons on kyasanur forest disease virus in cell culture. PLoS Negl. Trop. Dis. 2016, 10, e0004871. [Google Scholar] [CrossRef] [PubMed]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in west nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds trim25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Schnettler, E.; Sterken, M.G.; Leung, J.Y.; Metz, S.W.; Geertsema, C.; Goldbach, R.W.; Vlak, J.M.; Kohl, A.; Khromykh, A.A.; Pijlman, G.P. Noncoding flavivirus rna displays RNA interference suppressor activity in insect and mammalian cells. J. Virol. 2012, 86, 13486–13500. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindqvist, R.; Upadhyay, A.; Överby, A.K. Tick-Borne Flaviviruses and the Type I Interferon Response. Viruses 2018, 10, 340. https://doi.org/10.3390/v10070340
Lindqvist R, Upadhyay A, Överby AK. Tick-Borne Flaviviruses and the Type I Interferon Response. Viruses. 2018; 10(7):340. https://doi.org/10.3390/v10070340
Chicago/Turabian StyleLindqvist, Richard, Arunkumar Upadhyay, and Anna K. Överby. 2018. "Tick-Borne Flaviviruses and the Type I Interferon Response" Viruses 10, no. 7: 340. https://doi.org/10.3390/v10070340
APA StyleLindqvist, R., Upadhyay, A., & Överby, A. K. (2018). Tick-Borne Flaviviruses and the Type I Interferon Response. Viruses, 10(7), 340. https://doi.org/10.3390/v10070340