The HIV-1 Reverse Transcriptase A62V Mutation Influences Replication Fidelity and Viral Fitness in the Context of Multi-Drug-Resistant Mutations



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
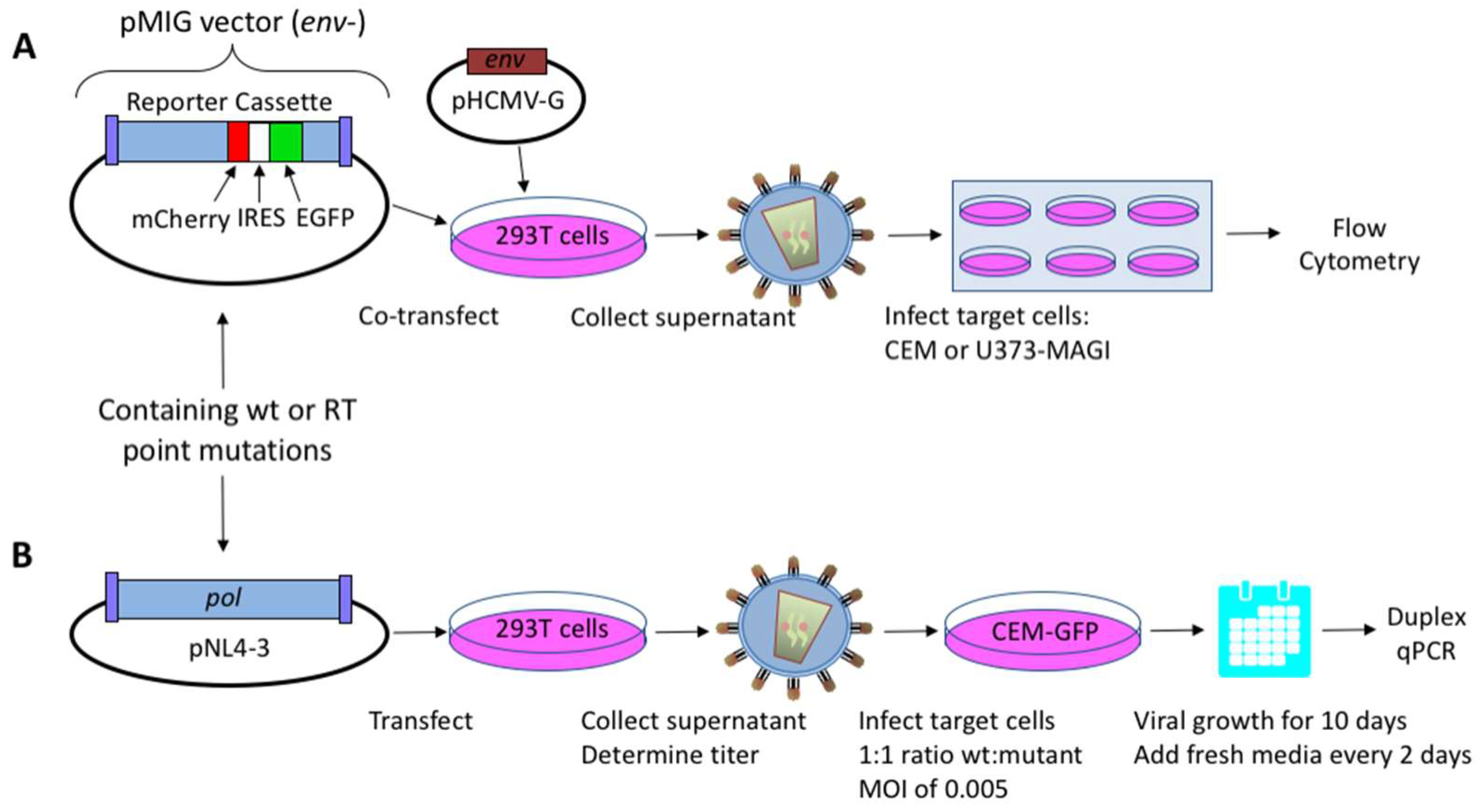
2. Materials and Methods
2.1. HIV-1 Vectors and Cell Lines
2.2. Virus Production and Titer Assay
2.3. Mutant Frequency Analysis by Flow Cytometry
2.4. Dual-Competition Assay
2.5. TaqMan Duplex qPCR Assay
2.6. Calculation of Viral Fitness
2.7. Statistical Analyses
3. Results
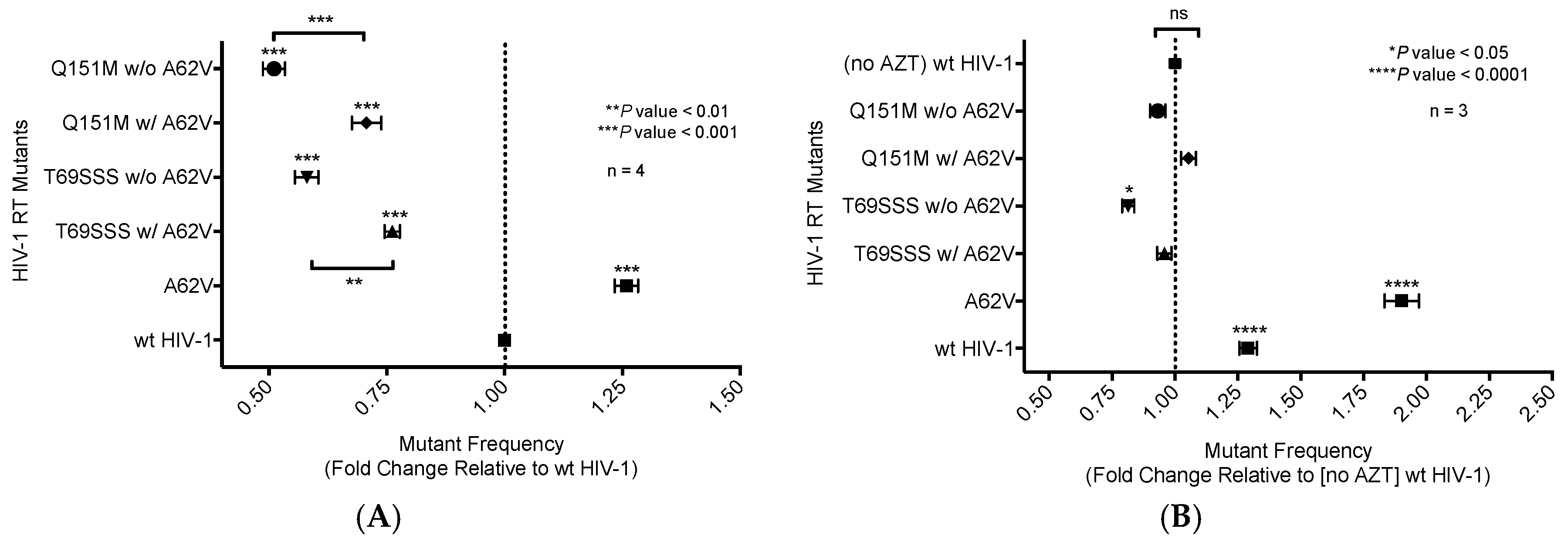
3.1. Mutant Frequency Analysis of HIV-1 RT Variants
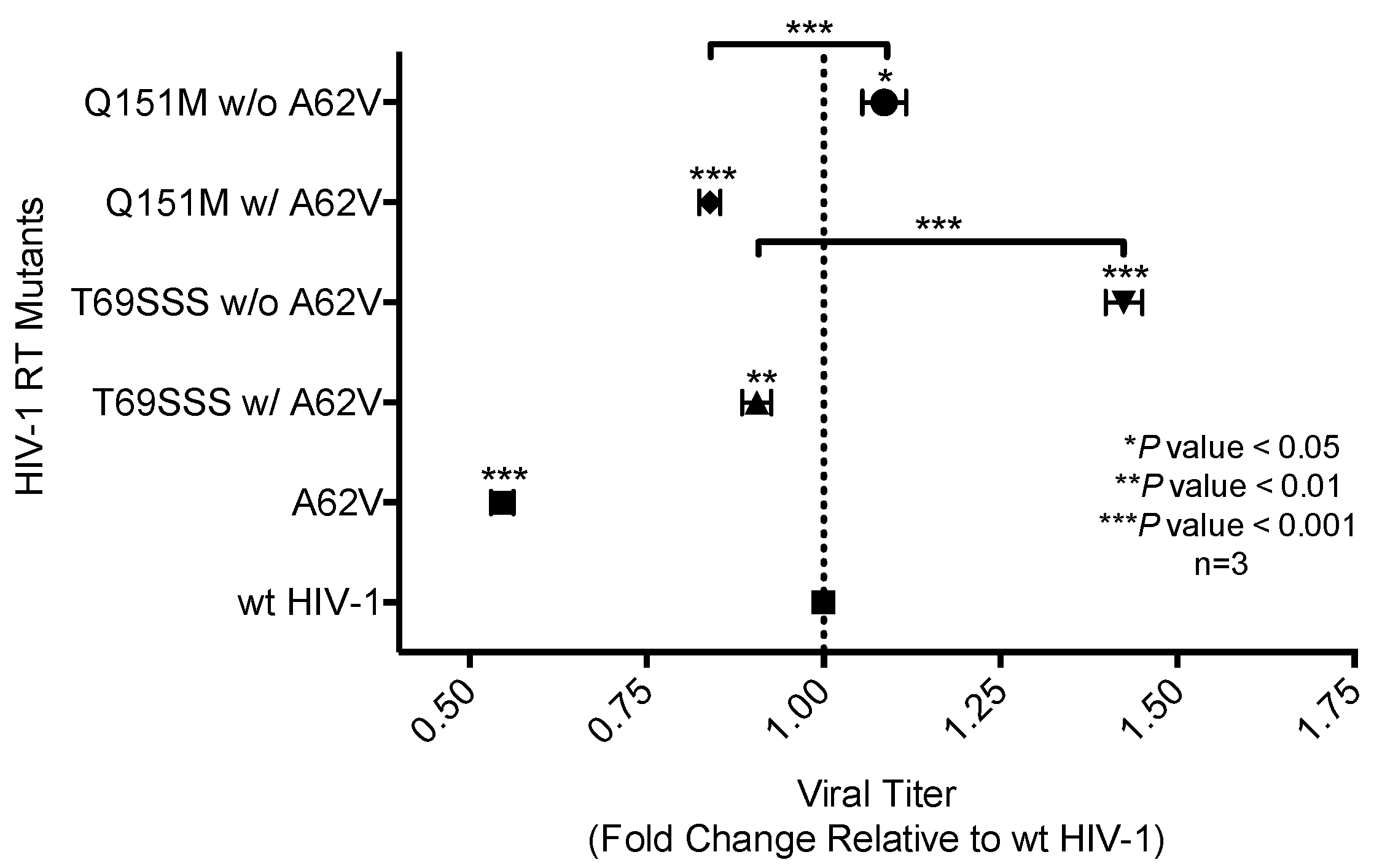
3.2. Replication Capacity Analysis of HIV-1 RT Variants
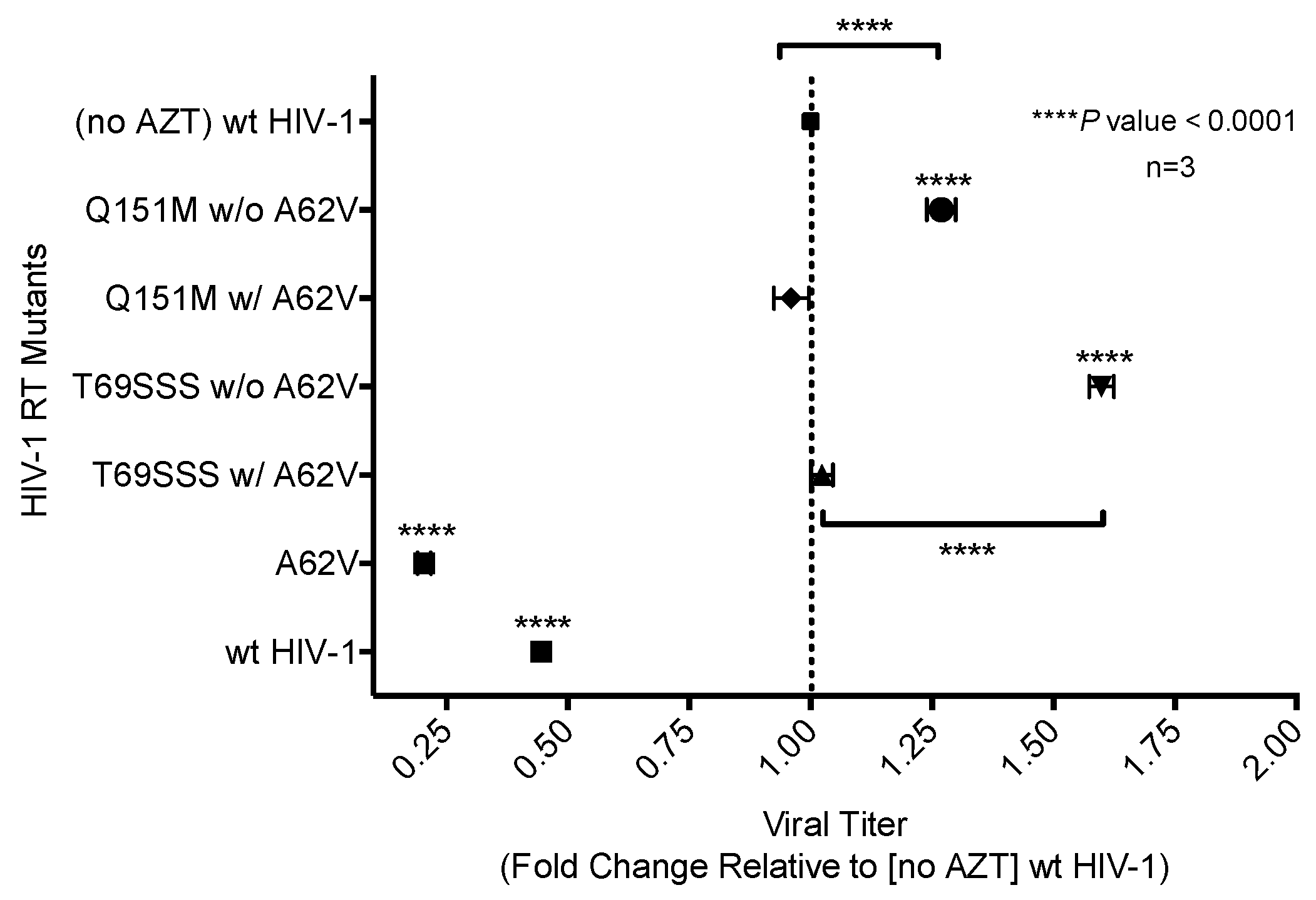
3.3. Drug Susceptibility of HIV-1 RT Variants to AZT
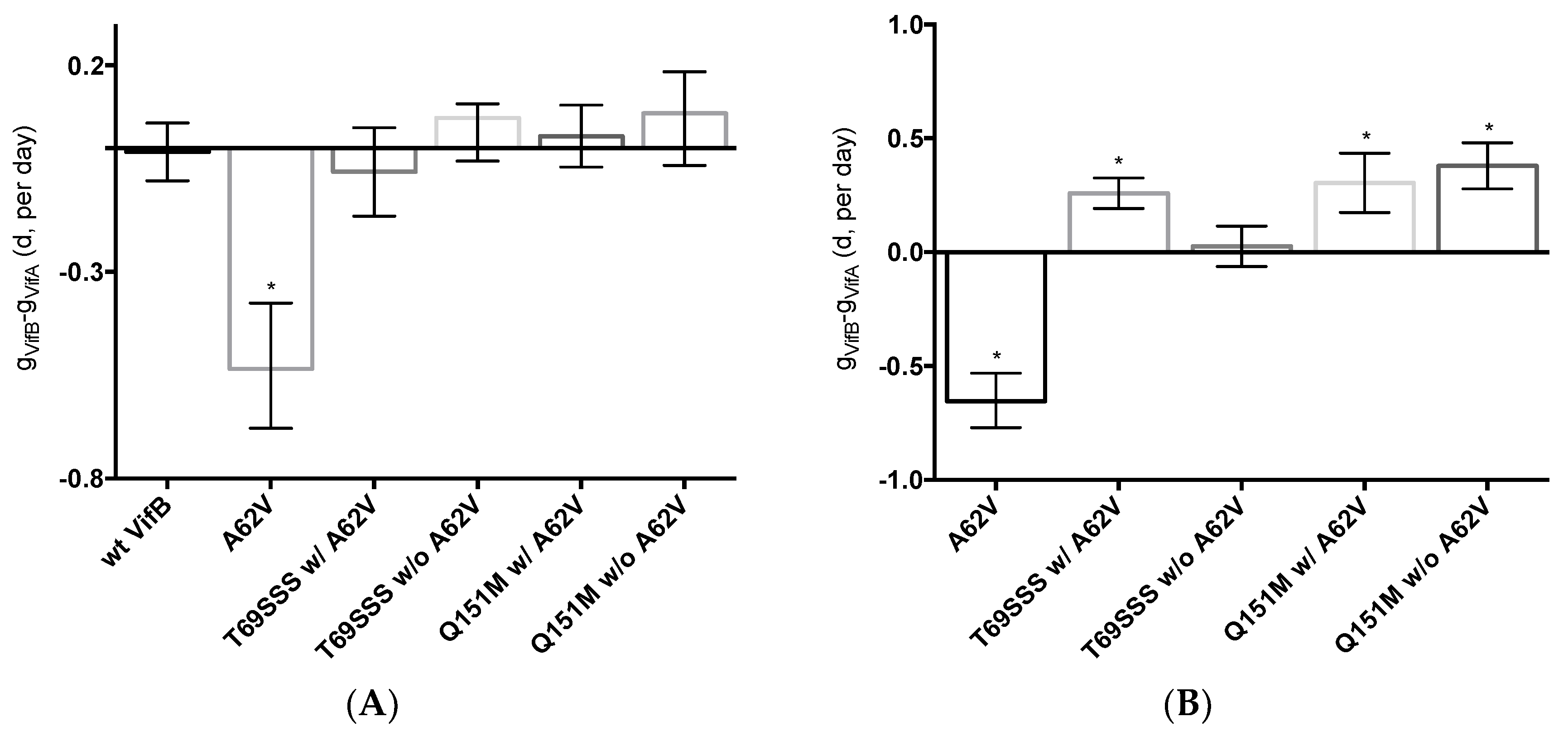
3.4. Fitness Impact of HIV-1 RT Mutations
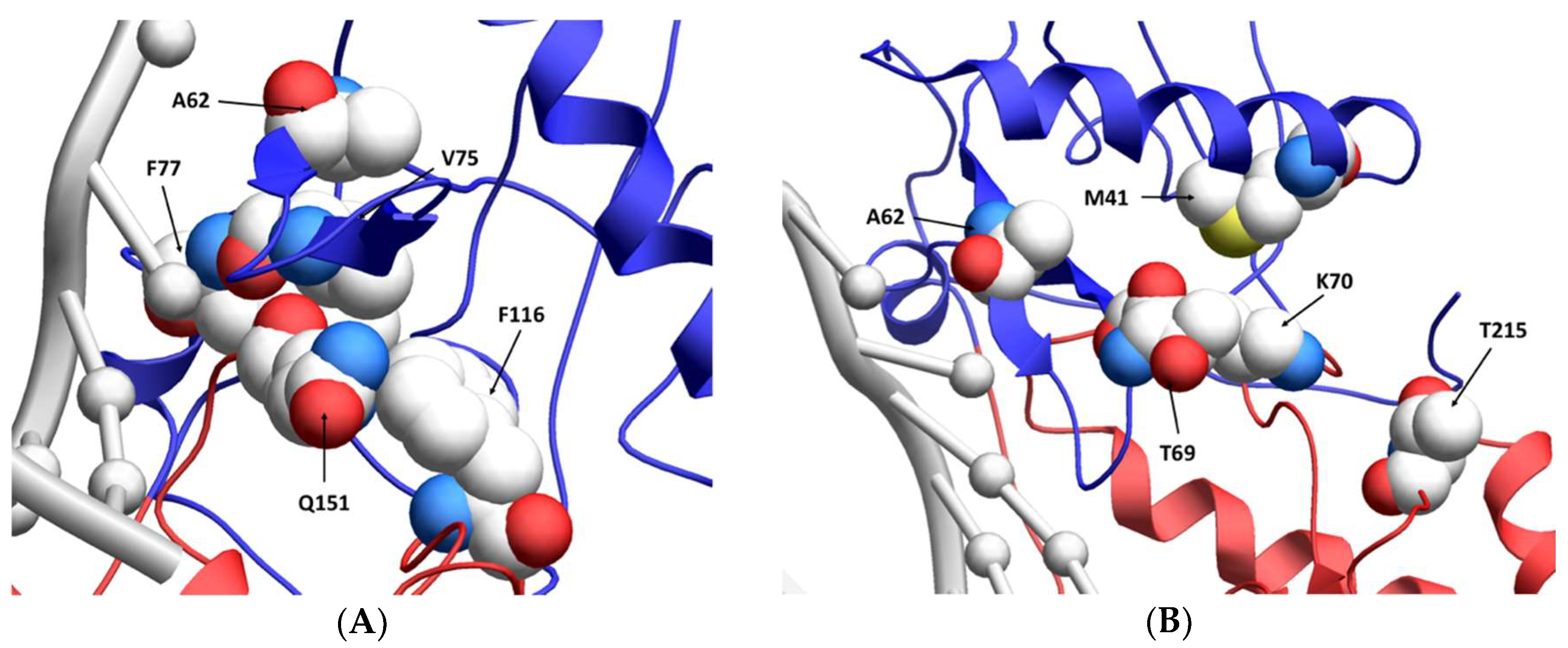
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, L.F.; Hoy, J.; Lewin, S.R. Ten years of highly active antiretroviral therapy for HIV infection. Med. J. Aust. 2007, 186, 146–151. [Google Scholar] [PubMed]
- Sluis-Cremer, N.; Arion, D.; Parniak, M.A. Molecular mechanisms of HIV-1 resistance to nucleoside reverse transcriptase inhibitors (NRTIs). Cell. Mol. Life Sci. 2000, 57, 1408–1422. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Non-nucleoside reverse transcriptase inhibitors (NNRTIs): Past, present, and future. Chem. Biodivers. 2004, 1, 44–64. [Google Scholar] [CrossRef] [PubMed]
- Detels, R.; Muñoz, A.; McFarlane, G.; Kingsley, L.A.; Margolick, J.B.; Giorgi, J.; Schrager, L.K.; Phair, J.P. Effectiveness of potent antiretroviral therapy on time to AIDS and death in men with known HIV infection duration. Multicenter AIDS Cohort Study Investigators. JAMA 1998, 280, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.L.; Collier, A.C.; Kalish, L.A.; Assmann, S.F.; Para, M.F.; Flanigan, T.P.; Kumar, P.N.; Mintz, L.; Wallach, F.R.; Nemo, G.J.; et al. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann. Intern. Med. 2001, 135, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yokoyama, M.; Sato, H.; Reilly, C.; Mansky, L.M. Human immunodeficiency virus mutagenesis during antiviral therapy: Impact of drug-resistant reverse transcriptase and nucleoside and nonnucleoside reverse transcriptase inhibitors on human immunodeficiency virus type 1 mutation frequencies. J. Virol. 2005, 79, 12045–12057. [Google Scholar] [CrossRef] [PubMed]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [PubMed]
- Coffin, J. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Escarmís, C.; Sevilla, N.; Moya, A.; Elena, S.F.; Quer, J.; Novella, I.S.; Holland, J.J. Basic concepts in RNA virus evolution. FASEB J. 1996, 10, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Clavel, F.; Race, E.; Mammano, F. HIV drug resistance and viral fitness. Adv. Pharmacol. 2000, 49, 41–66. [Google Scholar] [PubMed]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef] [PubMed]
- Persaud, D.; Zhou, Y.; Siliciano, J.M.; Siliciano, R.F. Latency in human immunodeficiency virus type 1 infection: No easy answers. J. Virol. 2003, 77, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, T.; Kavlick, M.F.; Ueno, T.; Gao, W.Y.; Kojima, E.; Alcaide, M.L.; Chokekijchai, S.; Roy, B.M.; Arnold, E.; Yarchoan, R. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. USA 1995, 92, 2398–2402. [Google Scholar] [CrossRef] [PubMed]
- Shafer, R.W.; Kozal, M.J.; Winters, M.A.; Iversen, A.K.N.; Katzenstein, D.A.; Ragni, M.V.; Meyer, W.A.; Gupta, P.; Rasheed, S.; Coombs, R.; et al. Combination therapy with zidovudine and didanosine selects for drug-resistant human immunodeficiency virus type 1 strains with unique patterns of pol gene mutations. J. Infect. Dis. 1994, 169, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, T.; Franco, S.; Vazquez-Alvarez, B.M.; Mas, A.; Martinez, M.A.; Menendez-Arias, L. Molecular determinants of multi-nucleoside analogue resistance in HIV-1 reverse transcriptases containing a dipeptide insertion in the fingers subdomain: Effect of mutations D67N and T215Y on removal of thymidine nucleotide analogues from blocked DNA primers. J. Biol. Chem. 2004, 279, 24569–24577. [Google Scholar] [PubMed]
- Menendez-Arias, L.; Matamoros, T.; Cases-Gonzalez, C.E. Insertions and deletions in HIV-1 reverse transcriptase: Consequences for drug resistance and viral fitness. Curr. Pharm. Des. 2006, 12, 1811–1825. [Google Scholar] [CrossRef] [PubMed]
- Cases-Gonzalez, C.E.; Franco, S.; Martínez, M.Á.; Menéndez-arias, L. Mutational patterns associated with the 69 insertion complex in multi-drug-resistant HIV-1 reverse transcriptase that confer increased excision activity and high-level resistance to zidovudine. J. Mol. Biol. 2007, 365, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Mas, A.; Parera, M.; Briones, C.; Soriano, V.; Martínez, M.A.; Domingo, E.; Menéndez-Arias, L. Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J. 2000, 19, 5752–5761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, A.K.; Shafer, R.W.; Wehrly, K.; Winters, M.A.; Mullins, J.I.; Chesebro, B.; Merigan, T.C. Multidrug-resistant human immunodeficiency virus type 1 strains resulting from combination antiretroviral therapy. J. Virol. 1996, 70, 1086–1090. [Google Scholar] [PubMed]
- Scherrer, A.U.; von Wyl, V.; Joos, B.; Klimkait, T.; Burgisser, P.; Yerly, S.; Boni, J.; Ledergerber, B.; Gunthard, H.F. Predictors for the emergence of the 2 multi-nucleoside/nucleotide resistance mutations 69 insertion and Q151M and their impact on clinical outcome in the Swiss HIV cohort study. J. Infect. Dis. 2011, 203, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Thuret, I.; Solas, C.; Genot, S.; Colson, P.; Tamalet, C. Vertical transmission of multidrug-resistant Q151M human immunodeficiency virus type 1 strains. Pediatr. Infect. Dis. J. 2008, 27, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.J.; Heineman, R.H.; Mansky, L.M. Interrelationship between HIV-1 fitness and mutation rate. J. Mol. Biol. 2013, 425, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.M.; Heineman, R.H.; Beach, L.B.; Martin, J.L.; Schnettler, E.K.; Dapp, M.J.; Patterson, S.E.; Mansky, L.M. 5,6-Dihydro-5-aza-2′-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorg. Med. Chem. 2013, 21, 7222–7228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, J.M.; Clouser, C.L.; Mansky, L.M. Rapid Determination of HIV-1 Mutant Frequencies and Mutation Spectra Using an mCherry/EGFP Dual-Reporter Viral Vector. Methods Mol. Biol. 2016, 1354, 71–88. [Google Scholar] [PubMed]
- Anastassopoulou, C.G.; Marozsan, A.J.; Matet, A.; Snyder, A.D.; Arts, E.J.; Kuhmann, S.E.; Moore, J.P. Escape of HIV-1 from a small molecule CCR5 inhibitor is not associated with a fitness loss. PLoS Pathog. 2007, 3, e79. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, M.A.; Goh, W.C.; Wu, L.I.; Rogel, M.E.; Bartz, S.R.; Schweickart, V.L.; Raport, C.J.; Emerman, M. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology 1997, 233, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Gervaix, A.; West, D.; Leoni, L.M.; Richman, D.D.; Wong-Staal, F.; Corbeil, J. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. USA 1997, 94, 4653–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Dapp, M.J.; Bonnac, L.; Patterson, S.E.; Mansky, L.M. Discovery of novel ribonucleoside analogs with activity against human immunodeficiency virus type 1. J. Virol. 2014, 88, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Holte, S.; Rao, U.; McClure, J.; Konopa, P.; Swain, J.V.; Lanxon-Cookson, E.; Kim, M.; Chen, L.; Mullins, J.I. A sensitive real-time PCR based assay to estimate the impact of amino acid substitutions on the competitive replication fitness of human immunodeficiency virus type 1 in cell culture. J. Virol. Methods 2013, 189, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhou, Y.; Alcock, C.; Kiefer, T.; Monie, D.; Siliciano, J.; Li, Q.; Pham, P.; Cofrancesco, J.; Persaud, D.; et al. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J. Virol. 2004, 78, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Venzon, D.J.; Mitsuya, H. Altered drug sensitivity, fitness, and evolution of human immunodeficiency virus type 1 with pol gene mutations conferring multi-dideoxynucleoside resistance. J. Infect. Dis. 1998, 177, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Kosalaraksa, P.; Kavlick, M.F.; Maroun, V.; Le, R.; Mitsuya, H. Comparative fitness of multi-dideoxynucleoside-resistant human immunodeficiency virus type 1 (HIV-1) in an In vitro competitive HIV-1 replication assay. J. Virol. 1999, 73, 5356–5363. [Google Scholar] [PubMed]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Shirasaka, T.; Mitsuya, H. Enzymatic characterization of human immunodeficiency virus type 1 reverse transcriptase resistant to multiple 2′,3′-dideoxynucleoside 5′-triphosphates. J. Biol. Chem. 1995, 270, 23605–23611. [Google Scholar] [CrossRef] [PubMed]
- Quinones-Mateu, M.E.; Tadele, M.; Parera, M.; Mas, A.; Weber, J.; Rangel, H.R.; Chakraborty, B.; Clotet, B.; Domingo, E.; Menendez-Arias, L.; et al. Insertions in the reverse transcriptase increase both drug resistance and viral fitness in a human immunodeficiency virus type 1 isolate harboring the multi-nucleoside reverse transcriptase inhibitor resistance 69 insertion complex mutation. J. Virol. 2002, 76, 10546–10552. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado, J.O.; Mansky, L.M. The HIV-1 Reverse Transcriptase A62V Mutation Influences Replication Fidelity and Viral Fitness in the Context of Multi-Drug-Resistant Mutations. Viruses 2018, 10, 376. https://doi.org/10.3390/v10070376
Maldonado JO, Mansky LM. The HIV-1 Reverse Transcriptase A62V Mutation Influences Replication Fidelity and Viral Fitness in the Context of Multi-Drug-Resistant Mutations. Viruses. 2018; 10(7):376. https://doi.org/10.3390/v10070376
Chicago/Turabian StyleMaldonado, José O., and Louis M. Mansky. 2018. "The HIV-1 Reverse Transcriptase A62V Mutation Influences Replication Fidelity and Viral Fitness in the Context of Multi-Drug-Resistant Mutations" Viruses 10, no. 7: 376. https://doi.org/10.3390/v10070376
APA StyleMaldonado, J. O., & Mansky, L. M. (2018). The HIV-1 Reverse Transcriptase A62V Mutation Influences Replication Fidelity and Viral Fitness in the Context of Multi-Drug-Resistant Mutations. Viruses, 10(7), 376. https://doi.org/10.3390/v10070376