Induction of Oxidative DNA Damage in Bovine Herpesvirus 1 Infected Bovine Kidney Cells (MDBK Cells) and Human Tumor Cells (A549 Cells and U2OS Cells)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Antibodies and Reagents
2.3. Immunofluorescence Assay
2.4. Western Blotting Analysis
2.5. Quantification of mRNA by qRT-PCR
2.6. Comet Assay
2.7. Cellular ROS Assay
2.8. Cell Viability Assay
3. Results
3.1. BoHV-1 Infection of MDBK Cells Results in Increased Levels of DNA Damage
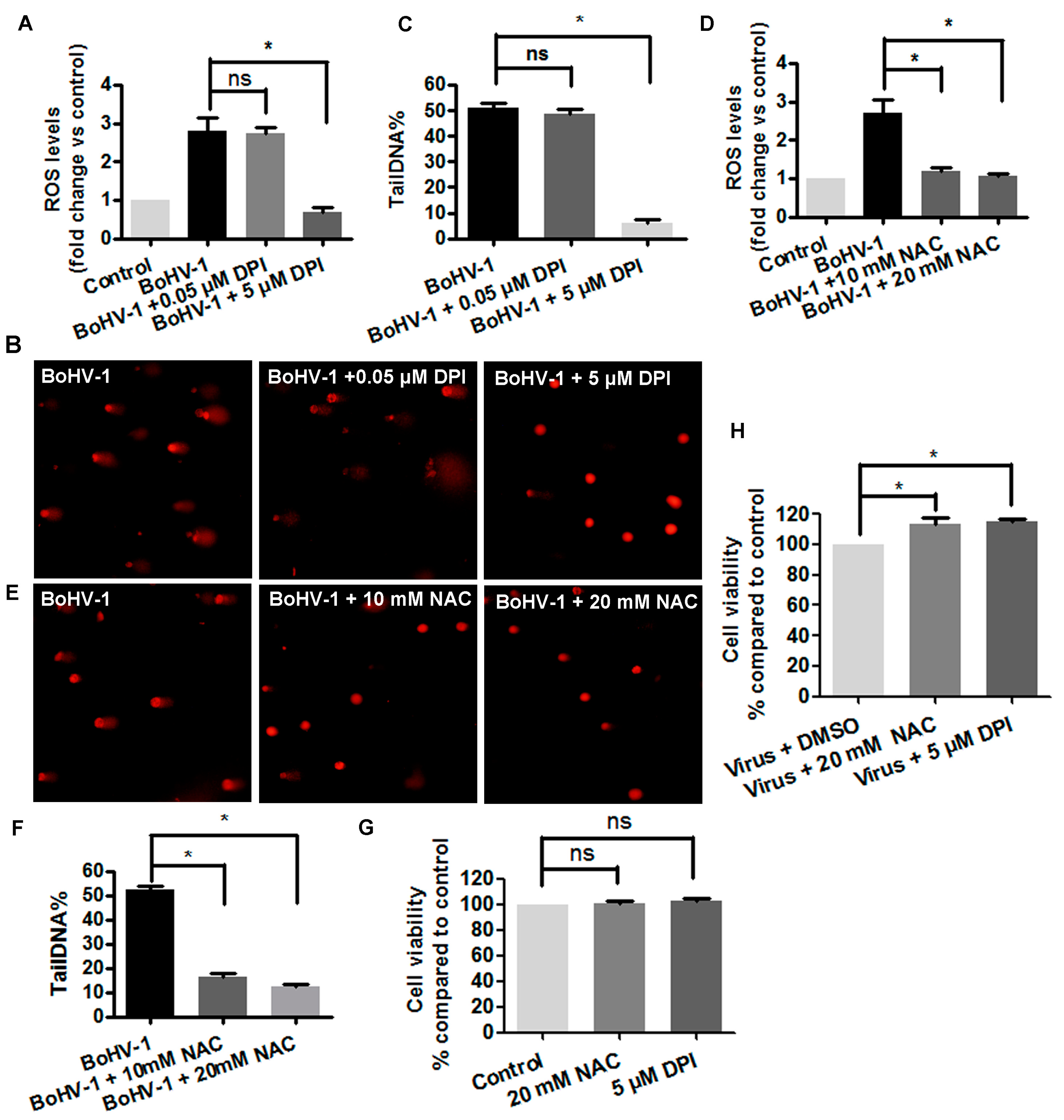
3.2. The Involvement of ROS in BoHV-1-Induced DNA Damages in MDBK Cells
3.3. BoHV-1 Infection of MDBK Cells Promotes 8-oxoG Production
3.4. ROS Levels Are Increased in BoHV-1 Infected U2OS Cells and A549 Cells
3.5. BoHV-1 Infection Leads to Elevated DNA Damage in Human Tumor Cells Such as U2OS Cells and A549 Cells
3.6. ROS Is Involved in BoHV-1-Induced DNA Damage in Human Tumor Cells
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Jones, C.; Chowdhury, S. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim. Health Res. Rev. 2007, 8, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Chase, C.C.L.; Fulton, R.W.; O’Toole, D.; Gillette, B.; Daly, R.F.; Perry, G.; Clement, T. Bovine herpesvirus 1 modified live virus vaccines for cattle reproduction: Balancing protection with undesired effects. Vet. Microbiol. 2017, 206, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.S.; Leunda, M.R.; Verna, A.E.; Moran, P.E.; Odeon, A.C.; Perez, S.E. Distribution of bovine herpesvirus type 1 in the nervous system of experimentally infected calves. Vet. J. 2016, 209, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, S.K.; Campos, M.; Babiuk, L.A. Bovine herpesvirus 1 (BHV-1): Biology, pathogenesis, and control. Adv. Virus Res. 1995, 45, 191–223. [Google Scholar] [PubMed]
- Srikumaran, S.; Kelling, C.L.; Ambagala, A. Immune evasion by pathogens of bovine respiratory disease complex. Anim. Health Res. Rev. 2007, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Vaha-Koskela, M.J.; Heikkila, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef] [PubMed]
- Willmon, C.; Harrington, K.; Kottke, T.; Prestwich, R.; Melcher, A.; Vile, R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, M.; Franceschi, V.; Capocefalo, A.; D’Avella, D.; Denaro, L.; Cavirani, S.; Mucignat-Caretta, C.; Donofrio, G. Herpes simplex virus type 1 thymidine kinase-armed bovine herpesvirus type 4-based vector displays enhanced oncolytic properties in immunocompetent orthotopic syngenic mouse and rat glioma models. Neuro-Oncology 2012, 14, 288–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddington, B.P.; Mossman, K.L. Oncolytic bovine herpesvirus type 1 as a broad spectrum cancer therapeutic. Curr. Opin. Virol. 2015, 13, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.; Cuddington, B.; Mossman, K. Bovine herpesvirus type 1 as a novel oncolytic virus. Cancer Gene Ther. 2010, 17, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Cuddington, B.P.; Verschoor, M.; Ashkar, A.; Mossman, K.L. Enhanced efficacy with azacytidine and oncolytic BHV-1 in a tolerized cotton rat model of breast adenocarcinoma. Mol. Ther. Oncolytics 2015, 2, 15004. [Google Scholar] [CrossRef] [PubMed]
- Cuddington, B.P.; Dyer, A.L.; Workenhe, S.T.; Mossman, K.L. Oncolytic bovine herpesvirus type 1 infects and kills breast tumor cells and breast cancer-initiating cells irrespective of tumor subtype. Cancer Gene Ther. 2013, 20, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Tapeinos, C.; Pandit, A. Physical, Chemical, and Biological Structures based on ROS-Sensitive Moieties that are Able to Respond to Oxidative Microenvironments. Adv. Mater. 2016, 28, 5553–5585. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sheng, W.S.; Schachtele, S.J.; Lokensgard, J.R. Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J. Neuroinflamm. 2011, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachtele, S.J.; Hu, S.; Little, M.R.; Lokensgard, J.R. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflamm. 2010, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, T.C.; Rosa, A.C.; Ferreira, H.L.; Okamura, L.H.; Oliveira, B.R.; Vieira, F.V.; Silva-Frade, C.; Gameiro, R.; Flores, E.F. Bovine herpesviruses induce different cell death forms in neuronal and glial-derived tumor cell cultures. J. Neurovirol. 2016, 22, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Jaime-Ramirez, A.C.; Bolyard, C.; Dai, H.; Nallanagulagari, T.; Wojton, J.; Hurwitz, B.S.; Relation, T.; Lee, T.J.; Lotze, M.T.; et al. Bortezomib Treatment Sensitizes Oncolytic HSV-1-Treated Tumors to NK Cell Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5265–5276. [Google Scholar] [CrossRef] [PubMed]
- Domingos, P.M.; Steller, H. Pathways regulating apoptosis during patterning and development. Curr. Opin. Genet. Dev. 2007, 17, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Caspase-dependent and -independent cell death pathways after DNA damage (Review). Oncol. Rep. 2005, 14, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Ditsworth, D.; Bauer, D.E.; Wang, Z.Q.; Thompson, C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004, 18, 1272–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 2006, 1757, 1371–1387. [Google Scholar] [CrossRef] [PubMed]
- Borges, H.L.; Linden, R.; Wang, J.Y. DNA damage-induced cell death: Lessons from the central nervous system. Cell Res. 2008, 18, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef] [PubMed]
- Misra, V.; Bratanich, A.C.; Carpenter, D.; O’Hare, P. Protein and DNA elements involved in transactivation of the promoter of the bovine herpesvirus (BHV) 1 IE-1 transcription unit by the BHV alpha gene trans-inducing factor. J. Virol. 1994, 68, 4898–4909. [Google Scholar] [PubMed]
- Shang, Y.; Zhang, L.; Jiang, Y.; Li, Y.; Lu, P. Airborne quinones induce cytotoxicity and DNA damage in human lung epithelial A549 cells: The role of reactive oxygen species. Chemosphere 2014, 100, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Fiorito, F.; Marfe, G.; De Blasio, E.; Granato, G.E.; Tafani, M.; De Martino, L.; Montagnaro, S.; Florio, S.; Pagnini, U. 2,3,7,8-tetrachlorodibenzo-p-dioxin regulates bovine herpesvirus type 1 induced apoptosis by modulating Bcl-2 family members. Apoptosis Int. J. Program. Cell Death 2008, 13, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, F.; Iovane, V.; Cantiello, A.; Marullo, A.; De Martino, L.; Iovane, G. MG-132 reduces virus release in Bovine herpesvirus-1 infection. Sci. Rep. 2017, 7, 13306. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, Y.; Costa, S.; Collins, A.R.; Azqueta, A. The comet assay, DNA damage, DNA repair and cytotoxicity: Hedgehogs are not always dead. Mutagenesis 2013, 28, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Jones, C. The high mobility group AT-hook 1 protein stimulates bovine herpesvirus 1 productive infection. Virus Res. 2017, 238, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.G.; Smith, J.A.; Balachandran, S.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Korth, M.J.; Barber, G.N.; Schiff, L.A.; Katze, M.G. The cellular protein P58IPK regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J. Virol. 2007, 81, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Becker, Y.; Asher, Y.; Cohen, Y.; Weinberg-Zahlering, E.; Shlomai, J. Phosphonoacetic acid-resistant mutants of herpes simplex virus: Effect of phosphonoacetic acid on virus replication and in vitro deoxyribonucleic acid synthesis in isolated nuclei. Antimicrob. Agents Chemother. 1977, 11, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yuan, C.; Zhang, D.; Ma, Y.; Ding, X.; Zhu, G. BHV-1 induced oxidative stress contributes to mitochondrial dysfunction in MDBK cells. Vet. Res. 2016, 47, 47. [Google Scholar] [CrossRef] [PubMed]
- Sova, H.; Jukkola-Vuorinen, A.; Puistola, U.; Kauppila, S.; Karihtala, P. 8-Hydroxydeoxyguanosine: A new potential independent prognostic factor in breast cancer. Br. J. Cancer 2010, 102, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [PubMed]
- Klungland, A.; Bjelland, S. Oxidative damage to purines in DNA: Role of mammalian Ogg1. DNA Repair 2007, 6, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.A.; Zharkov, D.O.; Grollman, A.P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 7429–7434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barghouth, P.G.; Thiruvalluvan, M.; LeGro, M.; Oviedo, N.J. DNA damage and tissue repair: What we can learn from planaria. Semin. Cell Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Park, E.M.; Lim, Y.S.; Hwang, S.B. Nonstructural 5A impairs DNA damage repair: Implication of hepatitis C virus-mediated hepatocarcinogenesis. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell. Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afroz, S.; Garg, R.; Fodje, M.; van Drunen Littel-van den Hurk, S. The major tegument protein of bovine herpesvirus-1, VP8, interacts with DNA damage response proteins and induces apoptosis. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Gay, C.M.; Byers, L.A. Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Transl. Lung Cancer Res. 2018, 7, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018, 66–67, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Alonso, M.M.; Gomez-Manzano, C.; Piao, Y.; Fueyo, J. Oncolytic viruses and DNA-repair machinery: Overcoming chemoresistance of gliomas. Expert Rev. Anticancer Ther. 2006, 6, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol. Cancer Ther. 2007, 6, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, V.; McFarlane, R.J.; Taylor, E.M.; Price, C. The genetics of the repair of 5-azacytidine-mediated DNA damage in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 1996, 251, 483–492. [Google Scholar] [PubMed]
- Kanai, R.; Rabkin, S.D.; Yip, S.; Sgubin, D.; Zaupa, C.M.; Hirose, Y.; Louis, D.N.; Wakimoto, H.; Martuza, R.L. Oncolytic virus-mediated manipulation of DNA damage responses: Synergy with chemotherapy in killing glioblastoma stem cells. J. Natl. Cancer Inst. 2012, 104, 42–55. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Fu, X.; Yuan, C.; Jiang, X.; Zhang, G. Induction of Oxidative DNA Damage in Bovine Herpesvirus 1 Infected Bovine Kidney Cells (MDBK Cells) and Human Tumor Cells (A549 Cells and U2OS Cells). Viruses 2018, 10, 393. https://doi.org/10.3390/v10080393
Zhu L, Fu X, Yuan C, Jiang X, Zhang G. Induction of Oxidative DNA Damage in Bovine Herpesvirus 1 Infected Bovine Kidney Cells (MDBK Cells) and Human Tumor Cells (A549 Cells and U2OS Cells). Viruses. 2018; 10(8):393. https://doi.org/10.3390/v10080393
Chicago/Turabian StyleZhu, Liqian, Xiaotian Fu, Chen Yuan, Xinyi Jiang, and Gaiping Zhang. 2018. "Induction of Oxidative DNA Damage in Bovine Herpesvirus 1 Infected Bovine Kidney Cells (MDBK Cells) and Human Tumor Cells (A549 Cells and U2OS Cells)" Viruses 10, no. 8: 393. https://doi.org/10.3390/v10080393
APA StyleZhu, L., Fu, X., Yuan, C., Jiang, X., & Zhang, G. (2018). Induction of Oxidative DNA Damage in Bovine Herpesvirus 1 Infected Bovine Kidney Cells (MDBK Cells) and Human Tumor Cells (A549 Cells and U2OS Cells). Viruses, 10(8), 393. https://doi.org/10.3390/v10080393